Thousands of hours of Molecular Dynamics saves you minutes of a Monte Carlo calculation
It seems that we have reached some kind of tipping point where microsecond molecular dynamics simulations are feasible. I'm starting to see many papers published where the only genuine point of interest seems to be that a protein was simulated for MORE THAN A MICROSECOND.
Still a microsecond is not quite long enough to watch biological stuff shuffle through interesting conformations without specifically tuning the simulation. What remains hard is to find something interesting for which a microsecond of simulation can simulate adequately.
And so, one of the more important papers from the measurement side of things came out in 2008 from Bert de Groot's group, Recognition Dynamics Up to Microseconds Revealed from an RDC-Derived Ubiquitin Ensemble in Solution, which claims to be the first NMR experiment to measure the complete microsecond dynamics of every residue in a protein. In this case, ubiquitin. The measurements are in the form of S order parameter, a measure of the spin relaxation of N atoms along the backbone derived from the Nuclear Magnetic Response. These are measured from Residual Dipolar Couplings.
The S order parameter is somewhat ill-defined with respect to the protein conformation, so there is some lattitude in how to interpret it. de Groot and coworkers showed that if the S order is interpreted as simulation constraints then an MD simulation with these constraints produce the same variabiality as an ensemble of 46 different crystal structures of ubiquitin, which translates the S order parameter into a quasi-experimental RMSD measure, which they call the EROS RMSDf.

With complete microsecond dynamics measurements, we finally have a litmus test for those spanking mew microsecond MD simulations. Step up to the plate, D.E. Shaw and coworkers, using their state-of-the-art Molecular Dynamics package DESMOND: Microsecond Molecular Dynamics Simulation Shows Effect of Slow Loop Dynamics on Backbone Amide Order Parameters of Proteins. So how did they do? If we plot the calculated S order parameters with the ones derived from the Residual Dipolar Couplings (actually I plot 1-S as I find it easier to see), we get an okay match of r = 0.63:
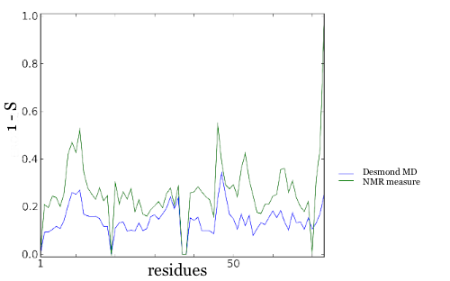
But here's the rub, in the course of my research into long time dynamics, I came across a whole bunch of great approximations that simulated protein flexibility (actually a reviewer suggested these to me). In particular, I was pretty impressed with CONCOORD, written by the very guy who made the NMR measurements.
The idea behind CONCOORD is simple and elegant: replace every bond, and inter-atomic interaction in a crystal structure with distance constraints that represent the strength of the interaction, and run a monte-carlo simulations. Using CONCOORD, in several minutes, I got a decent ensemble, from which I get the CONCOORD RMSDf. Comparing the experimentally derived EROS RMSDf to the CONCOORD RMSDf, I got a match r=0.54:

Comparing DESMOND to CONCOORD, the improvement of correlation to experiment was about 0.1 of correlation. The lesson to learn is that if you're running MD, you have to run it smart. The reason that CONCOORD performs well for ubiquitin, is that the motions of ubiquitin are actually not that large. Compared to the 6 or 7 Å motions of the ligand-binding loops of TIM or DHFR, ubiquitin really doesn't move all that much. Hence even a stripped down model such as CONCOORD can reproduce the experiment almost as well as DESMOND. To simulate these motions with MD, you need tens of microseconds of simulations.
Minutes of calculation with CONCOORD produces pretty much the same result as hundred of thousands of hours of CPU time with DESMOND. There's a lesson in there somewhere.