Structural alignment done right
Originally written 17/4/2009.
What is it to be alike? When you're talking about protein structures, you need to align the structures as best you can before you can talk about similarity. This is not a trivial matter. The standard approach is to use the Kabsch RMSD algorithm to find the optimal superposition between two sets of coordinates.
But if you've had any experience with real structures, or molecular dynamics trajectories, this is not a straightforward process. This is because some regions of the protein may vary much more than other regions. To get a better fit, you may need to apply "human intuition" to manually select a region that appear to be more stable, and then align the structures only to these regions, letting the rest flop away. Manually selecting the stable region is, to say the least, somewhat arbitrary.
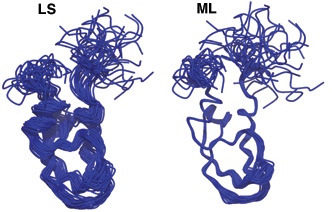
Except you don't need to fudge it anymore! Douglas Theobald and Deborah Wuttke have provided a new way of aligning structures THESEUS that automagically detects stable regions from variable regions, and aligns to the more stable regions, instead of the everything-in-the-blender of the Kabsch least-squares approach. It's a beautiful algorithm, and here's their example of the spectacularly better alignment of NMR models of the Kunitz domain 2sdf where LS is the Kabsch least-squares approach and ML refers to the Maximum Likehood approach of Theseus:
The killer application is in aligning crystal structures of different proteins in the same domain family. Typically, you first select a set of "conserved" residues and then do a Kabsch least-squares to just the "conserved" residues. This is normally an iterative process where you manually refine just which residues are conserved. Instead, Theseus hooks nicely into multiple-sequence alignment programs, and using the sequence alignment, it can produce a totally automated structual alignment of the structures that is every bit as good, if not better than a manual approach.
As an example, I've been working on the PDZ domains, an example 1x8s:

This structure has a canonical ligand-binding site running vertically in this viewing frame.
To make THESEUS easier to use, I have written PDBOVERLAY, to simplify
this kind of homology structural alignment. PDBOVERLAY is part of the
PDBREMIX package. Once installed,
this is how the alignment works for a bunch of PDZ domains, where
the -s flag means take only a single conformation from each PDB file:
> pdbfectch 1be9 1bfe 1d5g 1m5z 1nf3 1ry4 1rzx 1vj6 1x8s 2qku 3pdz
> pdboverlay -s *pdb
PDBOVERLAY uses MAFFT to align the sequences of the structures, THESEUS to do the alignment, and displays the structures using a special PYMOL script (blue:non-aligned-residues; green:non-protein-ligands):
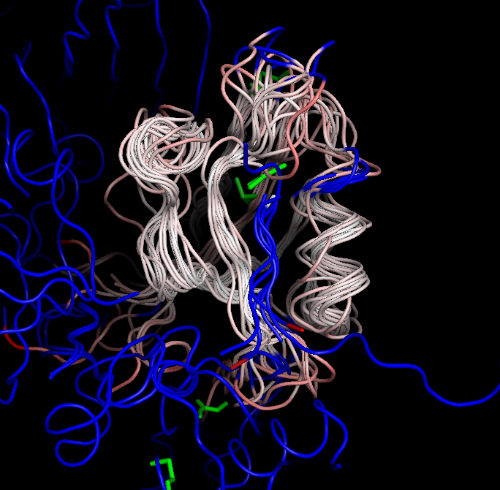
PDBOVERLAY can also align multiple models within a PDB file. For instance, 1ry4 is an NMR structure with 20 models, simply by running:
> pdboverlay 1ry4.pdb
The multiple models are automatically aligned by THESEUS to the best region:
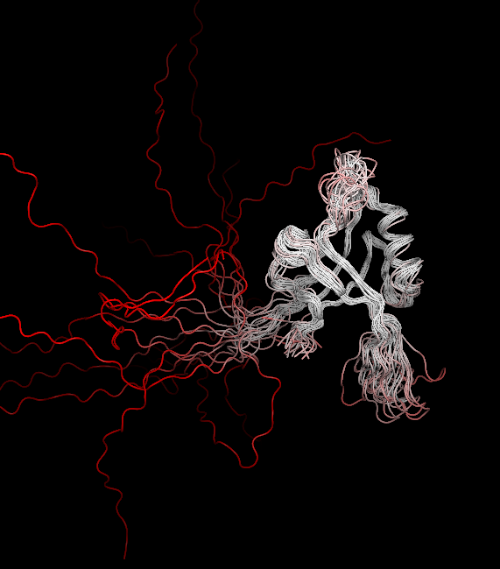
I never once had to apply "human intuition". Aligning structures has never been this easy!
PDBOVERLAY wraps THESEUS with MAFFT and PYMOL to make it easy to align and view homologous proteins and multiple models. It is part of PDBREMIX.