
This web-page was formatted with the Supplescroll javascript plugin.
The original article appeared in 1981, as pp. 167-339 in volume 34 of Advances in Protein Chemistry published by Academic Press. The volume was kept in print for over twenty years but is no longer available. Since it is still being actively cited and has still-applicable content as well as historical interest, we are now making it available electronically.
Once protein structures are solved they remain valid, and these pattern analyses were done conservatively; therefore almost nothing here has since been invalidated. However, the immense growth in structures solved since 1981 has shown new fold types and prompted new analyses. Short of the truly daunting task of a complete update the best solution seemed to be the addition of commentary as flagged notes (in green), to alert the reader to the most important newer information with brief explanations and references to follow further. With the addition of the notes, it is once again reasonable to send students to this reference for a detailed overview of protein structure.
Since the original manuscript predated our first word processor and the illustrations were hand drawn (by JSR) and then reproduced by high contrast optical photography (by DCR), putting the 173 pages and 109 multi- part figures into electronic form and cleaning them up was a very large task, done by Claudia J. Richardson with help from Dave Richardson. The new notes were written by Jane Richardson. The HTML pages were implemented by Claudia Richardson and Bryan Arendall with the final design of both HTML and PDF pages by Bryan Arendall.
We use several typographical cues to indicate source and status of document text. Original text is colored black, is of Times font-face (or a Serif font-face called for in your browser). Updates, as noted, are colored green, are presented in a sans-Serif type face, the font size is smaller than original text, and the added text appears in brackets. Outdated text sections are grayed out and left in place, they are not removed: the text is changed from black to gray, reduced in size, and preceded by "****". Figures were scanned from original prints at high resolution then resized to fit the web pages and the PDF documents. As web images are limited to 72dpi, it was necessary to increase the size of a few images to decrease pixelation. Literature references can be viewed in three ways: 1) a bibliography is available, 2) clicking on the in-line citation will open a new, smaller browser window showing the bibliography entry, and 3) hovering the mouse pointer over the in-line citation for a few seconds, will in most browsers bring up a text-box showing the bibliography entry. Document navigation can also be done in three ways: 1) a table of contents is available, 2) at top-right on every page is a gray box containing context-sensitive links, and 3) at bottom of every page are links to move to the next or previous pages. Printing from the web will yield pages with no navigational content and 150dpi images. We cannot however control the page breaks which will occur. For higher quality printouts, PDF formatted documents are available for download. Links are at the bottom of the page: typically, two PDFs will be available: one will be for the content of the current document (e.g. section 1a), the other for the entire section.
Some pages have active displays in the form of Java displays of kinemage files. Your browser will have to have "Java-enabled" in its setup preferences to see these displays. The availability of the active display will be marked by two small images: the first is a button to launch the display in a separate window; the second is a thumbnail of the initial state of the kinemage. In the future, we plan to add further commentary and more on-line graphics in this form. For examples and explanations, see our web site at http://kinemage.biochem.duke.edu/ ]
X-Ray crystallography is a technically sophisticated but conceptually simple-minded method with the great advantage that, to a first approximation, its results are independent of whatever preconceptions we bring to the task. This was very fortunate in the case of proteins, because it is unlikely that we could ever have successfully made the jump to such elegant and complex structures as those shown in Figs. 1 and 2 if we had been obliged to rely on more logical and indirect methods. For small inorganic and organic molecules indirect inference had succeeded magnificently, so that X-ray crystallography provided no startling revelations but only a prettier and more accurate picture of what was already known. However, even after knowing what the answer should look like for proteins, 20 years of effort has failed to derive three-dimensional protein structures from spectroscopic and chemical data or from theoretical calculations.
[Even after more than 40 years, we have made significant progress but still have certainly not solved the problem of predicting structure from sequence. The biggest change has been homology modeling: if a structure is known for a related sequence, which is now increasingly likely, then an approximate structure can be built which is useful for many purposes. De novo prediction is now sometimes quite close, but is certainly not reliable or routine. The prediction effort has been enhanced, and can be followed, through the Critical Assessment of Structure Prediction (CASP) competition (e.g., (Tramontano and Morea, 2003) .]
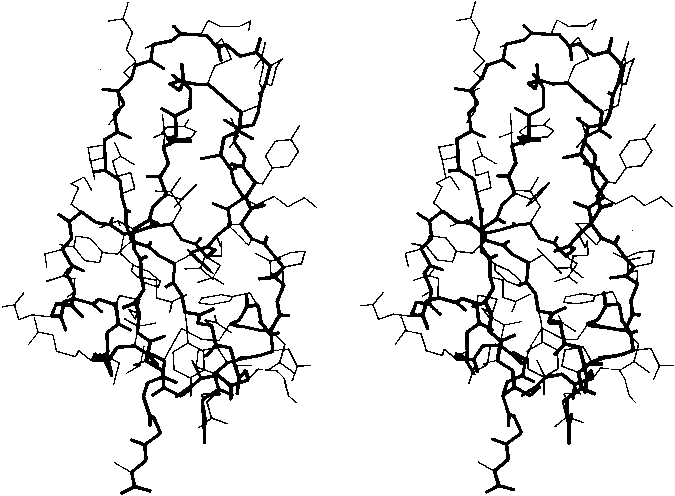
Before the first X-ray results, protein structure was visualized in terms of analogies based on chemistry and mathematics. The models proposed were relatively simple and extremely regular, such as geometrical lattice cages (Wrinch, 1937), repeating zigzags (Astbury and Bell, 1941), and uniform arrays of parallel rods (Perutz, 1949). In light of these very reasonable expectations, the low-resolution X-ray structure of myoglobin (Kendrew et al., 1958) came as a considerable shock. Kendrew, in describing the low-resolution model (see Fig. 3), says "Perhaps the most remarkable features of the molecule are its complexity and its lack of symmetry. The arrangement seems to be almost totally lacking in the kind of regularities which one instinctively anticipates." Perutz was even more outspoken about his initial disappointment: "Could the search for ultimate truth really have revealed so hideous and visceral-looking an object?" (Perutz, 1964).

In the last 20 years we have learned to appreciate the aesthetic merits of protein structure, but it remains true that the most apt metaphors are biological ones. Low-resolution helical structures are indeed "visceral," and high-resolution electron-density maps (for instance, see Fig. 13) are like intricate, branched coral, intertwined but never touching. β sheets do not show a stiff repetitious regularity but flow in graceful, twisting curves, and even the α-helix is regular more in the manner of a flower stem, whose branching nodes show the influences of environment, developmental history, and the evolution of each separate part to match its own idiosyncratic function.
The vast accumulation of information about protein structures provides a fresh opportunity to do descriptive natural history, as though we had been presented with the tropical jungles of a totally new planet. It is in the spirit of this new natural history that we will attempt to investigate the anatomy and taxonomy of protein structures.
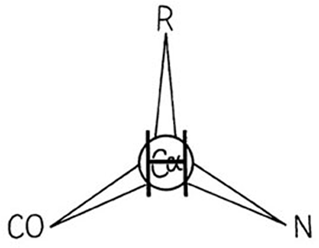
A protein, of course, is a polypeptide chain made up of amino acid residues linked together in a definite sequence. Amino acids are "handed" (except for glycine, in which the normally asymmetric α-carbon has two hydrogens), and naturally occurring proteins contain L-amino acids. That handedness has far-reaching effects on protein structure, as we shall see, and it is very useful to be able to distinguish the correct form easily. A simple mnemonic for that purpose is the "corncrib", illustrated in Fig. 4. Looking from the hydrogen direction, the other substituents around the α-carbon should read CO—R—N in a clockwise direction (R is the side chain). Threonine and isoleucine have handed β-carbons. A mnemonic for both of them is that if you are standing on the backbone with the hydrogen direction of the β-carbon behind you, then your left arm is the heavier of the two branches (the longer chain in Ile and the oxygen in Thr).
The sequence of side chains determines all that is unique about a particular protein, including its biological function and its specific three-dimensional structure. Each of the side groups has a certain "personality" which it contributes to this task. [see Richardson and Richardson (1989) for a more extensive discussion of amino-acid roles.] Histidine is the only side chain that titrates near physiological pH, making it especially useful for enzymatic reactions. Lys and Arg are normally positively charged and Asp and Glu negatively charged; those charges are very seldom buried in protein interiors except when they are serving some special purpose, as in the activity and activation of chymotrypsin (Blow et al., 1969; Wright, 1973). Asparagine and glutamine have interesting hydrogen-bonding properties, since they resemble the backbone peptides. The hydrophobic residues provide a very strong driving force for folding, through the indirect effect of their ceasing to disrupt the water structure once they are buried (Kauzmann, 1959); they also, however, affect the structure in a highly specific manner because their extremely varied sizes and shapes must all be fitted together in very efficient packing (Lee and Richards, 1971). Proline has stronger stereochemical constraints than any other residue, with only one instead of two variable backbone angles, and it lacks the normal backbone NH for hydrogen bonding. It is both disruptive to regular secondary structure and also good at forming turns in the polypeptide chain, so that in spite of its hydrophobicity it is usually found at the edge of the protein. Glycine has three different unique capabilities: as the smallest side group (only a hydrogen), it is often required where main chains must approach each other very closely; Gly can assume conformations normally forbidden by close contacts of the β-carbon; and it is more flexible than other residues, making it valuable for pieces of backbone that need to move or hinge.
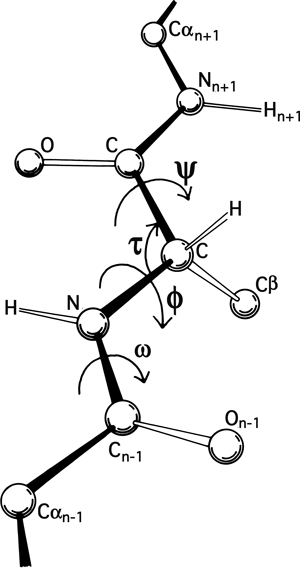
The basic geometry of amino acid residues is quite well determined from small-molecule crystal structures (see Momany et al., 1975; [also Engh and Huber, 1991; Engh and Huber, 2001] ). In terms of the accuracy of protein structure determinations, all of the bond lengths are invariant. Bond angles are also essentially invariant, except perhaps for τ, the backbone N—Cα—C angle (see Fig. 5). The α-carbon is tetrahedral, which would give 110°, but there are indications from accurately refined protein structures (e.g., Deisenhofer and Steigemann, 1975; Watenpaugh et al., 1979) that τ can sometimes stretch to larger values in order to accommodate other strains in the structure. [This has indeed been confirmed (e.g., Karplus, 1996). Other bond angles are also now standardly treated as variable, which is certainly realistic at least up to variations of 2-3° (Lovell et al., 2000).]
The dihedral angle ω at the peptide is very close to 180° (producing a trans, planar peptide with the neighboring α-carbons and the N, H, C, and O between them all lying in one plane), but there is evidence that ω can also vary slightly in real structures. Cis peptides, with ω = 0°, can occur perhaps 25% of the time in prolines [actually, more like 5-10% of the time] but essentially never for any other residue. The proline ring is not quite flat, and occasionally protein structures are now being refined accurately enough to determine the direction of ring pucker (e.g., Huber et al., 1974). [Ring puckers are now routinely assigned for Pro. The best evidence is that they should be either Cγ exo or Cγ endo (Némethy et al., 1992).] In the following discussions we will for the most part ignore possible effects such as proline ring pucker and variation in τ and ω.
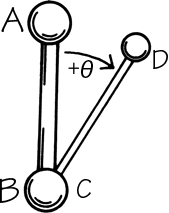
The remaining dihedral angles are the source of essentially all the interesting variability in protein conformation. As shown in Fig. 5 the backbone dihedral angles are φ and ψ in sequence order on either side of the α-carbon, so that φ is the dihedral angle around the N—Cα bond and ψ around the Cα—C bond. The side chain dihedral angles are χ1, χ2, etc. The four atoms needed to define each dihedral angle are taken either along the main backbone or out the side chain, in sequence order: N, Cα, C, N define ψ and N, Cα, Cβ, Cγ define χ1. The sign, or handedness, of any dihedral angle is defined as shown in Fig. 6: looking directly down the central bond (from either direction) and using the front bond as a stationary reference to define 0°, then the dihedral angle is positive if the rear bond is clockwise from 0° and negative if it is counterclockwise. The choice of reference atom (IUPAC-IUB, 1970) for side chain branches is made according to consistent chemical conventions, but it produces confusing results for the branched β-carbon residues since χ1 of 180° for Val puts its two Cγ atoms in the same position that the branches of Ile or Thr would occupy for χ1 = -60°.
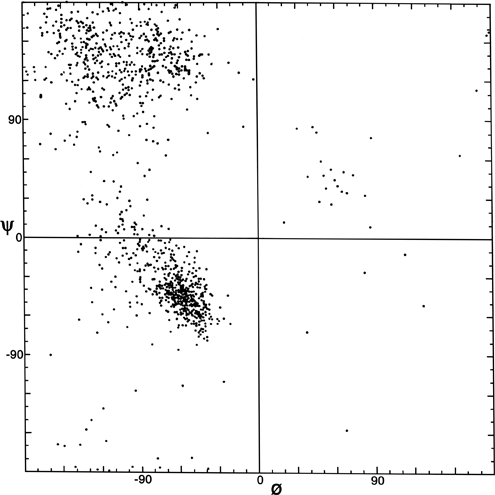
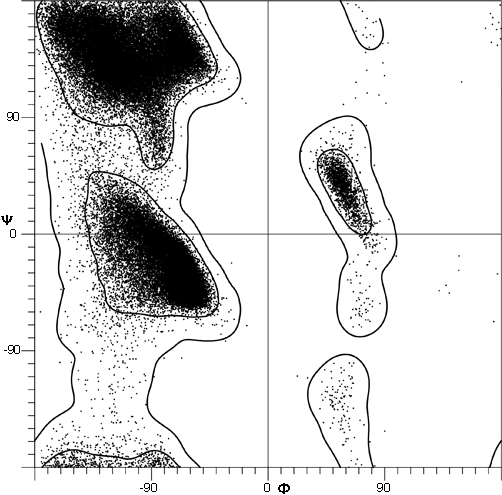
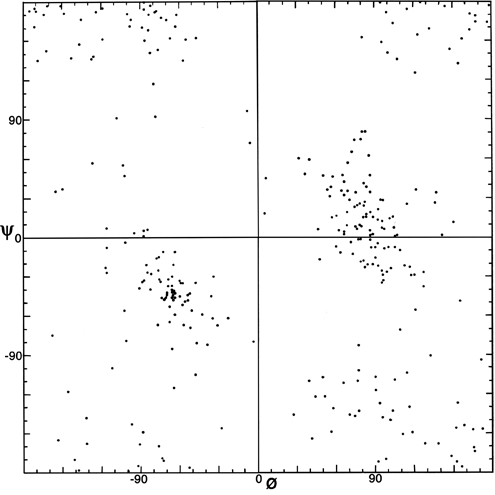
The parameters φ and ψ are the most important ones. An extremely useful device for studying protein conformation is the Ramachandran plot (Ramachandran et al., 1963) which plots φ and ψ. Figure 7 plots φ vs ψ for each nonglycine residue in eight of the most accurately determined protein structures (also picked to be representative of the various structure categories); Fig. 8 plots the glycine φ vs ψ from 20 proteins. The glycine plot is approximately symmetrical around the center, because glycine can adopt both right-handed and left-handed versions of any allowed conformation; however, there are some deviations from that symmetry, such as the different shapes and positions of the left- and right-handed α clusters. [Figures 7x and 8 give updated plots with more accurate and much larger datasets. Lovell et al. (2003) also show plots for the distinctly different cases of Pro and pre-Pro.]
***[Cautionary note: the conventions for naming and displaying φ and ψ have been changed twice. The original version in Ramachandran et al. (1963) defined ψ (called φ') in the same way as it is now used but defined φ as φ + 180°, so that the Ramachandran plot (with 0°,0° at the bottom left) had the α-helix in the upper left quadrant. Between 1966 and 1970, Ramachandran plots looked the same way they do now, but 0°,0° was at the bottom left and the numerical values of φ and ψ both differed by 180° from the current convention (e.g., Watson, 1969; Dickerson and Geis, 1969). Now 0°,0° is in the center of the φ,ψ plot, so that taking the mirror image of a conformation corresponds to inverting the numerical φ,ψ values through zero. For the current set of conventions, refer to the IUPAC-IUB Commission on Biochemical Nomenclature (1970) [and less drastic revisions that have appeared since (Markley et al., 1998;)] .]***
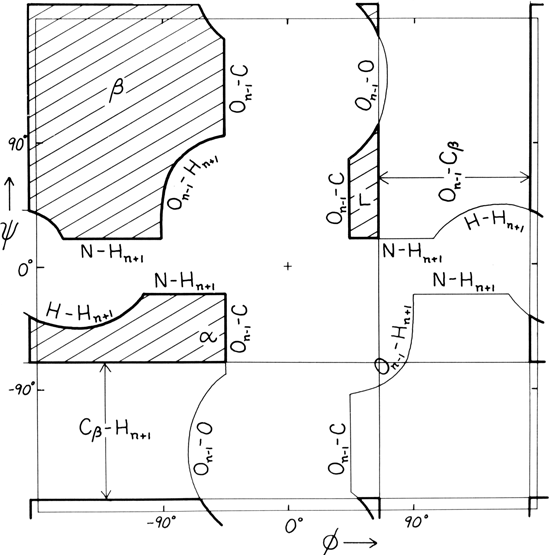
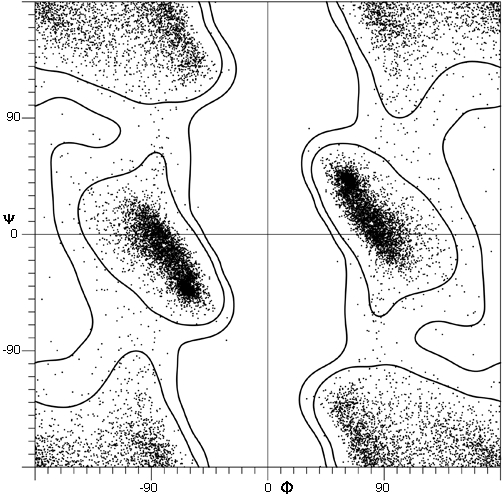
Theoretical calculations can provide a rather good understanding of these observed φ,ψ distributions. The first approach is to calculate what conformations are allowed without bump of hard sphere atoms of van der Waals radius. Figure 9 is a "derivation diagram" of the allowed regions, showing which pair of atoms is responsible for each forbidden zone (from Mandel et al., 1977). Four large regions symmetrical around 0°,0° are allowed for glycine. The presence of a β-carbon produces a bump with the carbonyl oxygen of residue n - 1 that is a function only of φ and not ψ and a bump with the NH of residue n + 1 that depends only on ψ and not φ. When the resulting vertical and horizontal disallowed strips are removed from the Ramachandran plot, one is left with fairly large regions around the β and the right-handed α conformations and a small region of left-handed α (Fig. 9). This outline fits the distribution observed in proteins (Fig. 7) fairly well, except for the rather frequent occurrence of residues in the bridge between the α and β regions. That bridge region becomes allowed if the C—Cα—N bond angle τ at the α-carbon is increased (e.g., Ramachandran and Sasisekharan, 1968), or if the grazing bump between N(i) and H(i+1) is otherwise softened. Detailed conformational energy calculations for alanine dipeptides (e.g., Maigret et al., 1971; Zimmerman and Scheraga, 1977a) can reproduce the observed distribution in most respects, in spite of omission of all long-range and medium-range interactions. [The shapes, heights, and positions of peaks and the outlines of favored regions on the updated Ramachandran plots of Fig. 7x and 8 are only approximately reproduced by Fig. 9y the dipeptide calculations referenced. Compare, for instance, the diagonal sharp edge of the α peak seen in Fig. 7xhe absence of data points in the predicted extension left of α. Significantly better agreement can be produced however, even for Gly, by quantum calculations (Hu et al., 2003).]
[Protein sidechains have between 0 and 4 χ dihedral angles (e.g., 0 for Gly, 1 for Ser, 2 for Phe, 3 for Met, 4 for Arg). χ angles around a bond joining two tetrahedral carbons have well-defined optima at staggered values (+60°, 180°, -60°); if one of the atoms has planar bonding (e.g., the Cγ of an aromatic ring), then the χ preferences are more complex. A very powerful generalization about sidechain conformations is that they are "rotameric" (Ponder and Richards, 1987) - that is, they populate well-defined clusters in the appropriate multi-dimensional space, corresponding to local energy minima. Rotamer "libraries" that list those favored conformation for each amino acid (Lovell et al., 2000; Dunbrack et al., 2002), or smoothed empirical probabilities in multidimensional χ space (MolProbity site at http://kinemage.biochem.duke.edu), are now used extensively in model buildiing of sidechains for either experimental or predicted protein structures.]
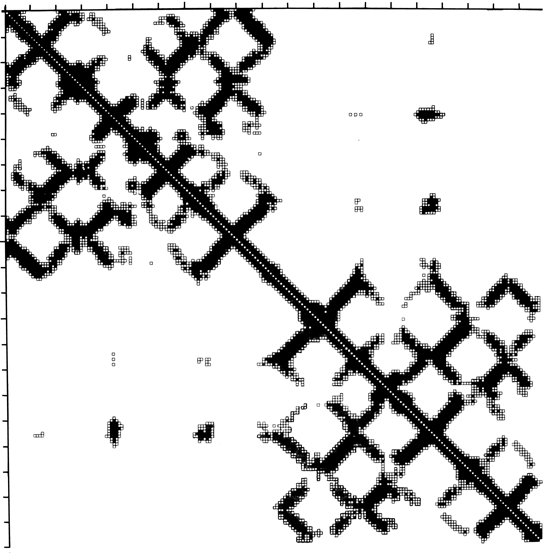
Another useful type of representation for protein structures is the diagonal plot. It is a matrix with the amino acid sequence number along both axes, in which either distance between the respective α-carbons or contact between the respective residues is plotted for each possible pair of residues (see Fig. 10). The diagonal plot is probably the most successful method yet devised of quantitatively mapping the chain folding in three dimensions onto the plane (stereo drawings are neither rigorously two-dimensional nor explicitly quantitative).
The large-scale structural features (except for handedness, and perhaps twist) have their counterpart in the diagonal plot: a helix gives a pronounced thickening along the diagonal, for instance, and a pair of antiparallel β strands produce a narrow stripe perpendicular to the diagonal. The appearance of each of the major structure types discussed in Sections III,B-E is fairly clear on diagonal plots, although less distinctive than in three dimensions. For example, the division into two well-separated domains with similar internal structures is extremely obvious in Fig. 10; the first layer of squares out from the diagonal indicates antiparallel organization (fairly narrow bands for β structure, as in this case, and wider bands if the elements were α-helices), the strong bands in the second layer are produced by the Greek key topology (see Section III,B), and the third layer is produced by closure of the barrel. Diagonal plot representations provide crucial simplifications of a number of computational problems (e.g., Kuntz et al., 1976; Tanaka and Scheraga, 1977; Remington and Matthews, 1978), and they seem to be an especially useful tool for those people who are more at home with an algebraic than with a geometrical representation.
[The most durably useful form of diagonal plot has turned out to be the "difference distance" diagonal plot which shows how different the distances are between the relevant atom pairs, for two different conformations or structures. ( Holm and Sander, 1993) It gives a detailed view of structural differences without need for superpositions.]
The following analysis and discussion of protein structure is based almost exclusively on the results of three-dimensional X-ray crystallography of globular proteins. In addition, one structure is included that was determined by electron diffraction (purple membrane protein), and occasional reference is made to particularly relevant results from other experimental techniques or from theoretical calculations. [More recently, large numbers of structures have also been contributed by nuclear magnetic resonance (NMR) methods, which use experimental measurements of distances between atoms and are reported as ensembles of multiple models equally consistent with the NMR data (see Cavanaugh et al., 2006). The Protein Data Bank (PDB) repository (Berman et al., 2002) passed 20,000 structures in 2002 and 40,000 in 2006; see http://www.rcsb.org/pdb. References to newer structures in the green update comments will use PDB IDcodes: 4 characters starting with a number, such as 1A6M for oxy-myoglobin at 1 Å resolution (Vojtechovsky et al., 1999).] Even with this deliberately restricted viewpoint the total amount of information involved is immense. Millions of independent parameters have been determined by protein crystallography, and the relationships among almost any subset of them are of potential interest. A major aim of the present study is to provide a guide map for use in exploring this forest of information.
One issue which needs to be discussed before starting the analysis is the problem of evaluating levels of probable error. X-Ray crystallography has a relatively high degree of inherent reliability, because it basically amounts merely to obtaining a picture of the protein. Serious mistakes or experimental difficulties usually produce recognizably unintelligible garbage rather than misleading artifacts. However, there are many minor inaccuracies or problems of interpretation that can affect reliability of the final coordinates. Also, there is now an enormous difference in accuracy between the best and the worst-determined structures: increasing numbers of large proteins are being solved for which the ordered diffraction pattern may not extend beyond 3.5 Å, while on the other hand it is now not uncommon for a protein structure to receive exhaustive least-squares refinement out to 1.5Å resolution. [The contrasts have continued to increase, with many biologically-important large complexes at quite low resolution, but with synchrotron data at cryogenic temperatures allowing some protein structures to be determined at better than 1Å resolution. The kinemage below shows a small piece of a refined electron density map at 1Å resolution (the heme group in the 1A6M myoglobin), illustrating its high and unambiguous information content.]
The problem of valid error estimation has not yet been solved even for a given refinement technique, mainly because it is difficult to estimate the likelihood of occasional large mistakes in assigning starting coordinates which might not be correctible by refinement. There are now a few cases in which the same structure was independently refined by different methods from independently determined starting coordinates (e.g., Huber et al., 1974; and Chambers and Stroud, 1979, for trypsin), or where two subunits related by noncrystallographic symmetry were refined independently (e.g., Mandel et al., 1977, for cytochrome c), so that we may soon develop some empirically based error-estimation procedures. So far the main conclusions from such comparisons are that temperature factors are good indicators of relative error level within a structure and that the standard deviation between independent, well-refined structures is very small (perhaps 0.1 or 0.2Å) for at least 90 or 95% of the atoms, but there are occasional quite large disagreements (as much as several angstroms) that fall well outside the tail of the normal distribution for the smaller errors. [Those occasional large differences or local errors mean that documented rmsd's between independently solved structures of the same molecule are 0.5 - 0.8Å for cases at about 2Å resolution (Kleywegt 1999; Mowbray et al., 1999).] For well-refined structures, then, the temperature factor (called "B" ; [see explanatory note] ) is inversely proportional to the relative accuracy of a given atom, or group, position. In the extreme case, an atom that refined to the maximum allowed temperature factor or that was in zero electron density has an essentially undetermined position, and quite probably is actually disordered in the protein.
[The B-factor is an exponential term in the crystallographic Fourier transform equation that describes how fast an atom's scattering falls off with resolution; at that atom position in the map, a high B-factor means that the electron density is smeared out or low. Thermal motion increases B-factors, as can any kind of error, but in proteins high B-factors are primarily due to multiple local conformations (i.e., disorder). B-factors are given for each atom in crystallographic PDB files, and some graphics programs can display or query them easily. Kinemage of 3LZM T4 lysozyme has B-values in the point ID (shown when an atom is picked) and is colored by B-factor on a black-body scale with blue coldest and white hottest.]
In addition to the relative local error level, one must bear in mind that there is always a small but finite probability that the position is grossly wrong, even for an apparently well-determined group. This probability is almost vanishingly small for a structure refined at, say, 1.5Å to a residual of 15% [Actually, it has turned out that even at high resolution sidechains such as Asn, Thr, or Leu can sometimes be fit backwards, producing atom positions off by several Å (Lovell et al., 2000).] ,but if the residual were 25 or 30% or the data only went out to 3Å resolution, then the likelihood of occasional large errors is quite substantial. [At these resolutions entire loops are sometimes misplaced or β-strands fit out of register.]
There are also some general rules of thumb that can be used to guess at error levels in unrefined and lower resolution structures. A first fundamental problem is to judge when there might be mistakes in the chain tracing that involve incorrect connectivity of the backbone. In a survey of 47 independent chain tracings of novel proteins which have been either confirmed or disconfirmed by further evidence, all of the tracings at 2.5Å resolution or better were correct, whether the sequence was known or not. Below 3.5Å resolution the sequence is irrelevant; with luck, an occasional structure can be traced reliably if it is simple and helical (e.g., Hendrickson et al., 1975). For the resolution range between 2.5 and 3.5Å, knowledge of the sequence makes considerable difference: only 20% of the structures with known sequences had to be rearranged, while two-thirds of those without sequences had at least one connectivity change. [Note that it is now quite rare to have an unrefined structure, or one with an unknown sequence.] Placement of all the major structural features is correct even when connectivity is not. Assignment of secondary structure elements is apt to be conservative in initial structure reports, so that the helices and β strands initially cited are almost invariably confirmed but additional elements may be recognized later.
In structures for which complete coordinates have been determined but not refined, error levels can be estimated according to position in the protein and what parameter is in question. Quite uniformly, main chain atoms are located more exactly than side chains and interior side chains are better determined than exposed ones. In general, positional parameters are more reliably known than dihedral angles. Ring plane orientation is much easier to determine for Trp, Tyr, and Phe than for His, because the electron density for a five-membered ring is nearly round at lower than about 2 Å resolution. Some parameters are especially prone to an occasional large error. If the carbonyl oxygen showed up clearly in the electron density, then φ and ψ are determined accurately, but if the carbonyl oxygen was not visible, then the orientation of the peptide is quite uncertain: in many cases it can flip by 180° without affecting positions of the surrounding α-carbons and side chains to any noticeable degree. Peptide rotation that is approximately independent of the surrounding chain can be seen between type I and type II tight turns (see Fig. 30). Peptide rotation involves a coupled change of ψ(n) and φ(n+1) by equal and opposite amounts. There may occasionally be true disorder of a peptide orientation in the protein, as has been suggested by dynamic calculations for several external peptides in pancreatic trypsin inhibitor (McCammon et al., 1977). φ and ψ are generally less accurately known for glycine than for other residues, because the β-carbon is not present in the map to help determine conformation. Another parameter subject to occasional large ambiguities is χ1. It is not too unusual, for instance, for the side chain electron density of a valine to show definite elongation parallel to the backbone direction but with no clear indication to which side the β-carbon protrudes. Of the two possible χ1 values one is staggered and one is eclipsed. If the crystallographer picks the staggered χ1 value he greatly improves his chances of being correct, but he is undermining the validity of future attempts at empirical determination of χ1 distributions. When the β-carbon is unbranched, the electron density sometimes extends out straight with no indication of the elbow bend at Cβ, in which case χ1 is also difficult to determine.
[Although not discussed here, nucleic acid structures and protein/nucleic acid complexes are an increasingly important part of structural biology. In DNA and RNA crystal structures the bases and the phosphates can be located very accurately but the rest of the backbone is quite difficult, with too many rotatable angles per observable atom (Murray et al., 2003).]
[For NMR structures, levels of error can be estimated by the number of experimental restraints per residue, which plays a role more or less analogous to resolution; "NOE" distance restraints between residues distant in sequence are especially critical. NMR structures are usually reported as an ensemble of multiple models all in similar agreement with the data; the spread between those models shows relative accuracy, and/or mobility, of local parts of the structure, analogous to the crystallographic B-factor.]
In summary, there are three important generalizations about error estimation in protein crystallography. The first is that the level of information varies enormously as a function primarily of resolution, but also of sequence knowledge and extent of refinement. The second generalization is that no single item of information is completely immune from possible error. If the electron density map is available or indicators such as temperature factors are known from refinement, then it is possible to tell which parameters are most at risk. The third important generalization is that errors occur at a very low absolute rate: 95% of the reported information is completely accurate, and it represents a detailed and objective storehouse of knowledge with which all other studies of proteins must be reconciled.
The α-helix is the classic element of protein structure. A single α-helix can order as many as 35 residues whereas the longest β strands include only about 15 residues, and one helix can have more influence on the stability and organization of a protein than any other individual structure element. α-helices have had an immense influence on our understanding of protein structure because their regularity makes them the only feature readily amenable to theoretical analysis.
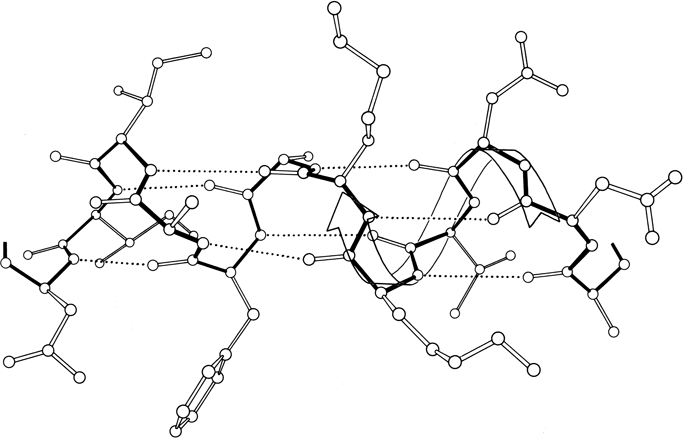
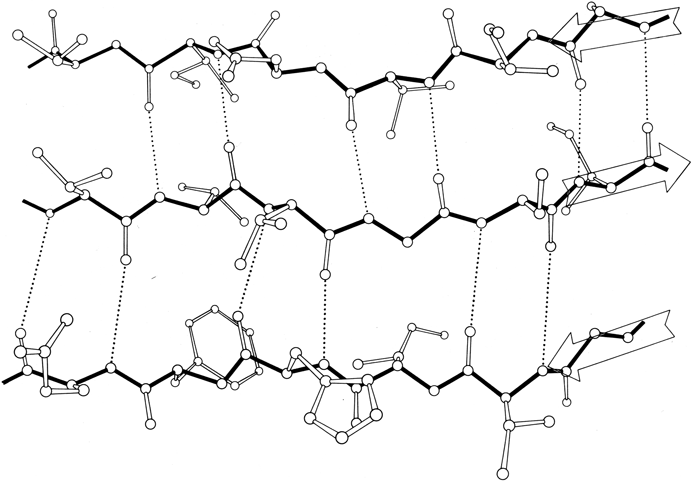
The α-helix was first described by Pauling in 1951 (Pauling et al., 1951) as a structure predicted to be stable and favorable on the basis of the accurate geometrical parameters he had recently derived for the peptide unit from small-molecule crystal structures. This provided the solution to the long-standing problem of explaining the strength and elasticity of the α-keratin structure and accounting for the appearance of its X-ray fiber diffraction pattern. Helices had frequently been proposed before as the α structure, but none of them could adequately match the diffraction pattern because they had been limited by the implicit assumption that a regular helix would necessarily have an integral number of amino acid residues per turn. In fact, as Pauling first realized, the α-helix has 3.6 residues per turn, with a hydrogen bond between the CO of residue n and the NH of residue n + 4 (see Fig. 11). The closed loop formed by one of these hydrogen bonds and the intervening stretch of backbone contains 13 atoms (including the hydrogen), as illustrated in Fig. 12. In the usual nomenclature for describing the basic structure of polypeptide helices, the α-helix is known as the 3.613-helix, where 3.6 is the number of residues per turn and 13 is the number of atoms in the hydrogen-bonded loop. The rise per residue along the helix axis is 1.5Å.
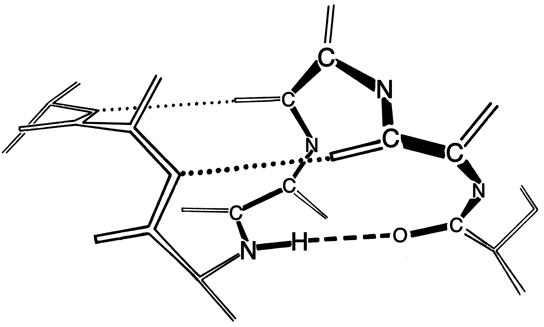
The α-helix received strong experimental support when Perutz (1951) found the predicted 1.5 Å X-ray reflection from hemoglobin crystals and from tilted fibers of keratins. The final conclusive demonstration of the α-helix in globular protein structure came from the high-resolution X-ray structure of myoglobin (Kendrew et al., 1960). It was shown that the myoglobin helices matched Pauling's calculated structure quite closely, and also that they were all right-handed (for L-amino acids, the left-handed α-helix has a close approach between the carbonyl oxygen and the β-carbon). It is easy to determine that, for instance, Fig. 11 is right-handed: if the curled fingers of the right hand are turned in the direction of their tips (as if tightening a screw) and the whole hand is moved in the direction of the outstretched thumb, then a right-handed helical path is traced out. Handedness is an enormously influential parameter in protein structure; most features for which handedness can be defined prefer one sense to the other, and the α-helix is only the first of many examples we will encounter.
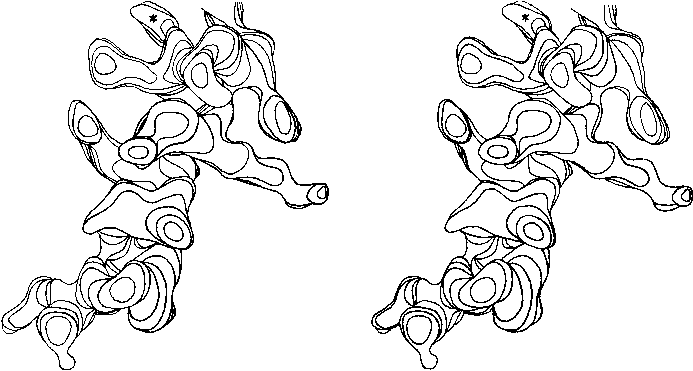
Figure 13 shows the electron density map at 2 Å resolution for one of the α-helices in staphylococcal nuclease. Bumps for the carbonyl oxygens are clearly visible; they point toward the C-terminal end of the helix, and are tipped very slightly outward away from the helix axis. At the top, in the last turn of the helix, there is a carbonyl tipped still further outward and hydrogen-bonded to a solvent molecule (marked with an asterisk). Side chain atoms or waters frequently bond to free backbone positions in the first or last turn of a helix, and hydrogen bonds with water are even more favorable for carbonyls than for NH groups (see Section II,H).
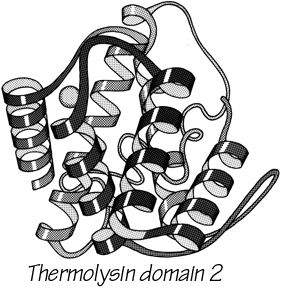
With 3.6 residues per turn, side chains protrude from the α-helix at about every 100° in azimuth. Since the commonest location for a helix is along the outside of the protein, there is a tendency for side chains to change from hydrophobic to hydrophilic with a periodicity of three to four residues (Schiffer and Edmundson, 1967). This trend can sometimes be seen in the sequence, but it is not strong enough for reliable prediction by itself. Different residues have weak but definite preferences either for or against being in α-helix: Ala, Glu, Leu, and Met are good helix formers while Pro, Gly, Tyr, and Ser are very poor (Levitt, 1977). α-Helices were central to all the early attempts to predict secondary structure from amino acid sequence (e.g., Davies, 1964; Guzzo, 1965; Prothero, 1966; Cook, 1967; Ptitsyn, 1969; Kotelchuk and Scheraga, 1969; Pain and Robson, 1970) and they are still the feature that can be predicted with greatest accuracy (e.g., Schulz et al., 1974b; Chou and Fasman, 1974b; Lim, 1974a; Matthews, 1975; Maxfield and Scheraga, 1976; Nagano, 1977b; Wu et al., 1978). [Helix predictions have now reached better than 70% accuracy, using algorithms such as neural nets (Rost and Sander, 2000) or hidden Markov models (Karplus et al., 1998).] As much as 80% of a structure can be helical, and only seven proteins are known that have no helix whatsoever. Figure 14 shows the second domain of thermolysin, a structure that is predominantly α-helical.
[There are of course now many more than 7 proteins known to have no helicies, but they are still a very small fraction of the total. Further information about amino-acid roles in helix formation is obtained from tabulating position-specific residue preferences (Richardson and Richardson, 1988). This shows that the ends of helices are very different from the central parts, as described below.]
The backbone conformational angles for right-handed α-helix are approximately φ= -60°, ψ= -60° [, more accurately, -63°, -43°] , which is in a favorable and relatively steep energy minimum for local conformation, even ignoring the hydrogen bonds. α-Helices are certainly the most regular pieces of structure to be found in globular proteins, but even so they show significant imperfections. There can be slight bends in the axis of a helix, of any amount from almost undetectable up to about 20° [30°] (e.g., Anderson et al., 1978), either with or without a break in the pattern of hydrogen bonding. One of the most obvious ways to produce such a bend is with a proline. Proline fits very well in the first turn of an α-helix [especially in position N1] but anywhere further on it not only is missing the hydrogen bond donor but also provides steric hindrance to the normal conformation. It is rare but certainly not unknown in such a position (see Fig. 15). An α-helix is almost invariably made up of a single, connected stretch of backbone (as opposed, for instance, to the backbone changeovers seen for double-helix in transfer RNAs: Holbrook et al., 1978). Almost the only known exception to this rule is the interrupted helix from subtilisin that is shown in Fig. 16.
[It has continued to prove true that strand changeovers are quite common in RNA molecules, with maintenance of base stacking and double helix geometry across the change, but interrupted α-helices with continuous H-bonding across the break remain extremely rare; one further example is in cytochrome C3 (1WAD 74-82), shown in the kinemage II.A_intHlx.kin.]
The generally regular, repeating conformation in the α-helix places all of the charge dipoles of the peptides pointing in the same direction along the helix axis (positive toward the N-terminal end). It has been shown (Hol et al., 1978) that the overall effect is indeed a significant net dipole for the helix, in spite of shielding effects. The helix dipole may contribute to the binding of charged species to the protein: for example, negative nucleotide phosphates, which are typically found near the N-termini of helices.
The only other principal helical species besides the α-helix which occurs to any great extent in globular protein structure is the 310-helix (see Fig. 17), with a three-residue repeat and a hydrogen bond to residue n + 3 instead of n + 4. Its backbone conformational angles are approximately φ = -60°, ψ = -30°, [-70°, -20°] , within the same energy minimum as the α-helix. However, for a long periodic structure the 310-helix is considerably less favorable than the α-helix in both local conformational energy and hydrogen bond configuration. In the refinement of rubredoxin at 1.2 Å resolution, Watenpaugh et al. (1979) found that bond angles along the main chain were significantly distorted in all four of the regions that have two successive 310-type hydrogen bonds. Long 310 helices are very rare but short pieces of approximate 310-helix occur fairly frequently. Two consecutive residues in 310 conformation form a good tight turn (see Section II,C), and three consecutive 310 residues forming two interlocked tight turns is also fairly common. But another important location for short bits of 310-helix is at the C-terminal end of an α-helix. It is quite common for the last helical turn to tighten up, with hydrogen bonds back to residue n - 3 or else bifurcated hydrogen bonds to both n - 3 and n - 4 (e.g., Watson, 1969). Némethy et al. (1967) showed that this arrangement is not necessarily quite like 310-helix; they described the αII-helix for this sort of position, which retains the helical parameters of an α-helix but tilts the peptide so that the NH points more inward toward the helix axis and at the same time points more toward the n - 3 than the n - 4 carbonyl. The conformations in real proteins show somewhat of a mixture between the αII tilt and the 310 tightening. Figure 18 shows an example. 310 or αII conformation does not tend to occur nearly as often at the N-termini of α-helices. The reason is that the tighter loop with n + 3-type hydrogen bonds requires the group involved to move closer to the helix axis, either by tilting (αII) or by tightening the helix(310). This motion is easy for the NH group but not for the CO: neighboring carbonyl oxygens would come too close together. [Less often, the end of a helix can loosen rather than tighten or a turn can widen to provide the right geometry for a metal ligand, using the (n+5) H-bonds of what is called a π-helix. An example is myohemerythrin 106-112 (2MHR).]
Another frequent feature of the C-termini of helices is a residue (usually glycine) in left-handed α conformation with its NH making a hydrogen bond to the CO of residue n - 5 (see Schellman, 1980); this often follows a residue with the 310 or αII bonding described above. [This arrangement has turned out to be very much the commonest way of ending an α-helix. The starting and ending residues that form the transition point half-in and half-out of a helix are now called the helix N-cap and C-cap respectively (Richardson, 1988). The C-cap is most often a glycine in L-α conformation that turns the backbone in the other direction; the peptides NH's on either side of the Gly Cα make H-bonds back to exposed CO's in the last helical turn, but in inverted sequence order (as shown in kinemage II.A_hlxCaps.kin for the Gly C-cap of helix 4 in 1LMB λ repressor). Helix N-cap residues usually have a short sidechain (Asn, Asp, Ser, or Thr) with an oxygen that can H-bond to the exposed backbone NH of residue N2 or N3 (that is, 2 or 3 past the N-cap) in the first helical turn (shown in kinemage for the N-caps of helices 1 and 2 of 1LMB λ repressor). A classic helix N-cap also has a "capbox" reciprocal H-bond from the sidechain of residue N3 (Gln, Glu, Ser, or Thr) to the backbone NH of the N-cap residue, in the peptide just before the start of helical conformation (Harper and Rose, 1993). Both N-caps and C-caps often also have a "hydrophobic staple" interaction between suitable sidechains at N' and N4 or C4 and C' (Muñoz et al., 1995). Proline is actually preferred in the N1 positions (Richardson, 1988).
Good N-caps stabilize both entire proteins (Serrano and Fersht, 1989; Nicholson et al., 1991) and isolated helical peptides (Lyu et al., 1993). Glycine C-caps do not stabilize helical peptides (Doig and Baldwin, 1995), but that has been shown to be due to their location at the C-terminus of the chain (Kapp et al., 2004). Sequences that form good helix caps have become important tools in secondary-structure prediction (Muñoz and Serrano, 1994) and in protein design (Marshall et al., 2002).]
A few other helical conformations occur occasionally in globular protein structures. The polyproline helix, of the same sort as one strand out of a collagen structure, has been found in pancreatic trypsin inhibitor (Huber et al., 1971) and in cytochrome c551 (Almassy and Dickerson, 1978). An extended "ε helix" has been described as occurring in chymotrypsin (Srinivasan et al., 1976). In view of the usual variability and irregularity seen in local protein conformation it is unclear that either of these last two helix types is reliably distinguishable from simply an isolated extended strand; however, the presence of prolines can justify the designation of polyproline helix.
[sidebar: Analyses of Helix-Helix Packing]
***The ways in which α-helices pack against one another were initially described by Crick (1953) as "knobs into holes" side chain packing which could work at either a shallow left-handed crossing angle or a steeper right-handed one. Helix-helix interactions have recently been analyzed in more detail by several different groups, using quite varied approaches and points of view. Chothia et al. (1977) considered the helix contact angles at which ridges formed by rows either of n,n + 3 or of n,n + 4 side chains can pack against each other. They predict three classes (I, II, and III) of contact at angles of -82°, -60° and +19°, respectively (the angle is handed but does not consider direction of the helices). For 25 cases they find a distribution consistent with these classes, although there is better discrimination between classes II and III than between I and II. Richmond and Richards (1978) determine contact residues by calculating solvent accessible area lost on bringing helix pairs together, and model the interactions using helices of close-packed spheres. They find contact classes that match the packing of Chothia's classes II and III, but for approximately perpendicular helices (class I) they find a favorable contact only if the two central residues are glycine or alanine and pack directly on top of each other. In globins the helix axes are about 2Å closer together for steeply angled contacts than for nearly parallel ones, which have a long contact surface between relatively large residues. Figure 19 shows stereo drawings of class II and class III helix contacts. Efimov (1977, 1979) also considers side chain packing as the determinant for helix contacts, but from a rather different theoretical perspective. He first considers what side chain conformations will allow close packing of neighboring hydrophobic side chains on a single helix, then considers how to close-pack side chains of hydrophobic patches on the buried side of two parallel or antiparallel helices, then finally considers the angles for packing together two layers of helices by matching two of the relatively flat hydrophobic surfaces produced in the second step.
Each of these approaches has its advantages; the contact nets drawn by Chothia et al. are the only version that explicitly shows the actual (rather than idealized) residue contacts, but they have made correlations only with the one variable of contact angle. Efimov has obtained a very interesting regularity that successfully predicts side chain conformation at the right and left edges of hydrophobic strips, but has not considered either the interactions directly in between helix pairs in his first step or the possibility that close (as opposed to distant hydrophobic) contacts could occur at steep angles. Richmond and Richards have the advantage of identifying residue contacts in a way that is not influenced by theoretical preconceptions, and they have considered side chain identity (although not conformation) in detail. Because of the great local variability of side chain size and packing and because relatively few examples have yet been analyzed, it is obviously possible to describe a given contact as fitting quite different idealized models. The current large data set of proteins shows a strong tendency for class III (shallow) interactions to be antiparallel and for parallel helix interactions to be class II. It seems likely that the antiparallel up and down helix bundle structures (see Section III,B) would be composed of paradigm class III interactions, and the doubly wound α/β structures (see Section III,C) would contain paradigm class II interactions, but none of the 15 proteins analyzed by the above three methods happen to fall into either of those categories. If multiple examples of paradigm classes II and III contacts can be analyzed and compared, it may then be possible to define a meaningfully distinct class of perpendicular contacts.***
[Analysis of the general geometry of helix packing is still a fairly open issue, but several aspects of the problem have progressed. An interesting treatment of helix packing in terms of alternate edges of polyhedra (Murzin and Finkelstein, 1988) fits well for many but not all structures. The common and biologically important case of coiled-coils (belonging to the low-angle, class III case) has been very thoroughly and successfully described (O'Shea et al., 1991), and a rare type of low-angle contact at much closer distances has been described (Gernert et al., 1995). Perpendicular T-junction contacts have proven important in Ca++-binding "E-F hands" (Kretsinger, 1980) and DNA-binding helix-turn-helix motifs (Steitz et al., 1982).]
The other major structural element found in globular proteins is the β sheet. Historically, it was first observed as the β, or extended, form of keratin fibers. An approximate understanding of the molecular structure involved was achieved much earlier for the β than for the α structure, because repeat distances along the fiber showed that the backbone must be almost fully extended, which did not leave very much choice of conformation even when the details of backbone geometry were not well known. Astbury described the β structure in 1933 as straight, extended chains with alternating side chain direction and hydrogen bonds between adjacent antiparallel chains. Pauling and Corey (1951) described the correct hydrogen-bonding patterns for both antiparallel and parallel β sheet, and also realized that the sheets were "pleated," with α-carbons successively a little above and below the plane of the sheet. Some features of β structure, such as its characteristic twist, were not recognized until after several β sheets had been seen in three-dimensional protein structures.
β sheet is made up of almost fully extended strands, with φ,ψ angles which fall within the wide, shallow energy minimum in the upper left quadrant of the Ramachandran plot (see Figs. 7 and 9). β strands can interact in either parallel or antiparallel orientation, and each of the two forms has a distinctive pattern of hydrogen bonding. Figures 20 and 21 illustrate examples of antiparallel and parallel β sheets from real protein structures. The antiparallel sheet has hydrogen bonds perpendicular to the strands, and narrowly spaced bond pairs alternate with widely spaced pairs. Looking from the N- to C-terminal direction along the strand, when the side chain points up the narrow pair of H bonds will point to the right. Parallel sheet has evenly spaced hydrogen bonds which angle across between the strands.
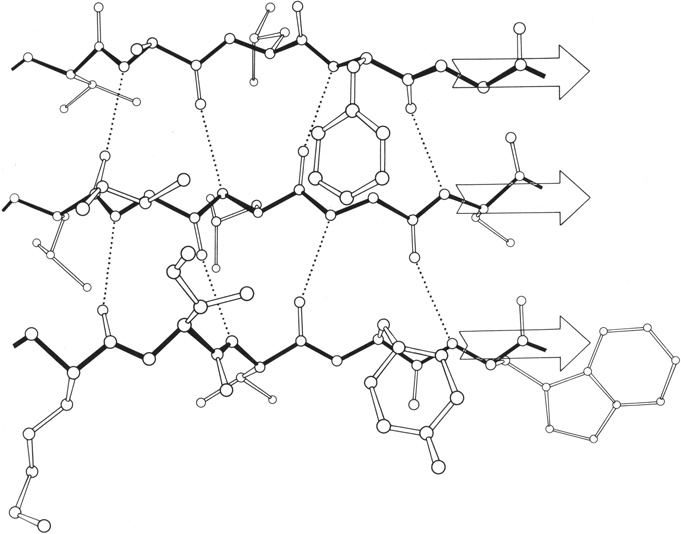
Within a β sheet, as within an α-helix, all possible backbone hydrogen bonds are formed. In both parallel and antiparallel β sheet, the side groups along each strand alternate above and below the sheet, while side groups opposite one another on neighboring strands extend to the same side of the sheet and are quite close together. These close side chain pairs on neighboring strands show preferences for having hydrophobic groups together, unlike charges together, and branched β-carbons next to unbranched β-carbons (in antiparallel sheet), but none of these preferences are stronger than 2 to 1. Lifson and Sander (1980a,b) have shown that specific residue pairs on neighboring strands recognize each other, over and above simple grouping by polarity, but again they comment on the fact that the correlations are not as strong as one would have expected. As an example of the kind of factors involved, let us examine the interactions of a pair of side chains with branched β-carbons on neighboring strands of β sheet. Valine and isoleucine have a rather strong conformational preference (better than two-thirds of the cases) for the χ1 orientation staggered relative to the main chain (Janin et al., 1978). Since the relation between adjacent parallel strands is a translation, neighboring Val or Ile residues in the preferred conformation "cup" against each other back-to-front in a very favorable packing. Since the relationship between adjacent antiparallel strands is twofold, in that case a pair of side chains with the preferred χ1 angle will either pack back-to-back leaving unfilled space or else front-to-front, which produces a collision unless the main chain conformation is adjusted. The effects of these restrictions can indeed be seen in the patterns of residue-pair occurrence, but only weakly. Looking at the actual pairs of, for instance, Val-Val or Val-Ile in antiparallel sheet, one finds either that one of the side chains has adopted an unfavorable χ1 angle so that the two can pack well (as in the upper left corner of Fig. 20) or else the main chain has twisted to put the β-carbons at an optimum distance (e.g., when a Leu-Val pair in chymotrypsin becomes a Val-Val pair in elastase, the β-carbons move 0.65 Å further apart). This in turn, of course, shows one reason why the χ1 preference is not stronger or the φ,ψ angles more regular. In general, the impression one takes away from this kind of examination is that the protein is balancing so many factors at the same time that there are always ways to compensate for any individual problem. Thus studies of individual parameters uncover only weak regularities in spite of the strength of the overall packing constraints. Looking at long strings of adjacent side chains across the centers of large sheets, such as shown in the stereo figures of Lifson and Sander (1980b), one sees a stronger expression of the packing difference between antiparallel and parallel sheets: Ile-Leu-Val-Leu and Val-Ala-Thr-Gly-Ile in elastase and Ala-Ile-Ala-Val, Ala-Ile-Leu-Ile-Ala, and Ser-Thr-His-Val-Ser in concanavalin A, versus Val-Val-Ile-Val-Val-Val and Ile-Val-Ile in glyceraldehyde-phosphate dehydrogenase domain 1 and Val-Val-Ile, Val-Val-Val, and Ile-Ile-Val in triosephosphate isomerase. [Wouters and Curmi (1995) give an updated statistical study of pair frequencies in β sheet, while the energetics of specific replacements have been studied experimentally, especially in the nicely behaved B domain of protein G (e.g., Smith et al. 1994).]
β strands can combine into either a pure parallel sheet, a pure antiparallel sheet, or a mixed sheet with some strand pairs parallel and some antiparallel. If the assortment were random there would be very few pure sheets, but in fact there is a strong bias against mixed sheets (Richardson, 1977), perhaps because the two types of hydrogen bonding need slightly different peptide orientations. Only about 20% of the strands inside β sheets have parallel bonding on one side and antiparallel on the other.
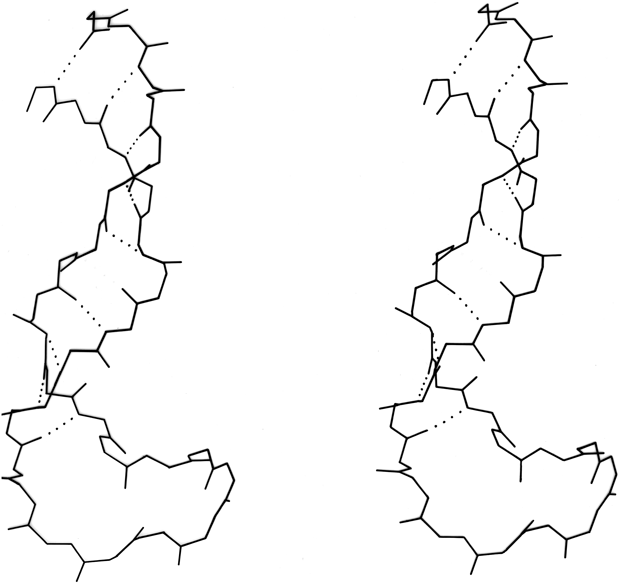
Parallel β sheet is in general a good deal more regular than antiparallel. If φ,ψ angles are plotted for both types of sheet, as for instance in Nagano (1977a), the parallel residues cluster rather tightly while the antiparallel ones spread over the entire quadrant. Parallel β structure almost never occurs in sheets of less than five total strands, whereas antiparallel β structure often occurs as a twisted ribbon of just two strands. Figure 22 shows such a two-stranded antiparallel β ribbon. Parallel β sheets and the parallel portions of mixed sheets are always thoroughly buried, with other main chain (often α-helices) protecting them on both sides. Antiparallel sheets, on the other hand, typically have one side exposed to solvent and the other side buried, so that they often show an alternation of side chain hydrophobicity in the amino acid sequence. [The contrast is still very real, but there are now examples of solvent-exposed parallel β sheet: on some parts of parallel β-helix structures such as LpxA (1LXA), or on the inner surface of α/β horseshoes such as ribonuclease inhibitor (1DFJ). This seems to happen, however, only for highly repetitive and very regular parallel β-sheets, and probably benefit from stabilization by their cooperativity.] β sheets in general show a tendency toward greater hydrophobicity for the central than for the edge strands of the sheet (Sternberg and Thornthon, 1977c). These three requirements of parallel β sheets (regularity, size, and protection) all suggest that parallel β structure is less stable than antiparallel (Richardson, 1977), since it apparently needs the cooperativity of an extensive hydrogen-bond network (see Sheridan et al., 1979) and also seems to need those hydrogen bonds shielded from water. (It is actually possible to shield the backbone with large hydrophobic side chains, but those are not the residues that would occur on an exposed surface.) Mixed β sheets tend to have the general appearance characteristic of their predominant H-bonding type. Sheets that are approximately half and half, such as carboxypeptidase or carbonic anhydrase, tend to look like parallel sheets because they require substantial protection on both sides. Figure 23 is a schematic drawing of a typical parallel-type β sheet structure in a protein.

One of the most conspicuous features of β sheet as it occurs in the known protein structures is its twist (Chothia, 1973). This twist always has the same handedness, although it has unfortunately been described by two conflicting conventions in the literature. If defined in terms of the angle at which neighboring β strands cross each other, then the twist is left-handed (e.g., Quiocho et al., 1977; Shaw and Muirhead, 1977); if defined in terms of the twist of the hydrogen bonding direction or of the peptide planes as viewed along a strand, then the twist is right-handed (e.g., Schulz et al., 1974a; Chothia et al., 1977). We will use the right-handed definition in this article, because it is meaningful even for an isolated strand. [The right-handed definition won out and is now standard.] Figure 23 shows the side view of a β sheet in which the twist is obvious.
There is of course no a priori reason to expect the flat n = 2 conformation to be especially favored for handed amino acids; however, the exact mechanism by which L-amino acids favor right-handed strand twist is not entirely obvious and has been explained in several different ways. Detailed calculations of local conformational energy (e.g., Zimmerman and Scheraga 1977a) always place the minimum well off to the right of the n = 2 line of a flat strand (see Dickerson and Geis, 1969) although the minimum is a very broad, shallow one. Chothia (1973) points out that probabilistic effects will produce a right-handed average twist, since many more of the accessible conformations within the general β area on the φ,ψ plot lie to the right of the n = 2 line. Raghavendra and Sasisekharan (1979) have found that inclusion of H bond and nonbonded interactions between a pair of antiparallel β strands produces a considerably deeper calculated energy minimum in the right-handed region. There is some evidence from small-molecule peptide crystal structures (Ramachandran, 1974) of a systematic tetrahedral distortion at the peptide nitrogen, and Weatherford and Salemme (1979) have shown that the combination of that distortion with optimal β sheet hydrogen bond geometry would favor a right-handed strand twist. In the known structures, β strand twist varies from close to 0° per residue to about 30° per residue, with the highest values for two-stranded ribbons (see Fig. 22) and generally lower values the more strands are present and the longer they are. This indicates some degree of conflict between the requirements for optimal hydrogen bonding and for lowest local conformational energy. [Indeed, if a large sheet were strongly twisted then the H-bonds would have to be longer at the edges; a greater degree of "pleat" at the sheet center helps somewhat, but flatness helps even more. These relationships were explored in Salemme (1983).]
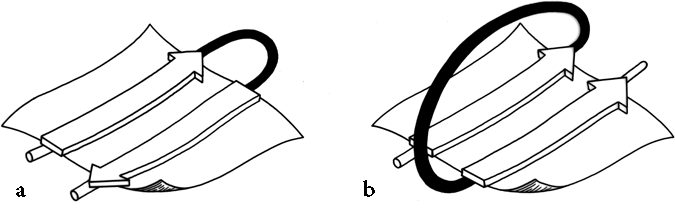
Once it has been decided what β strands belong in a given sheet (a process involving occasional subjective decisions for marginal cases), then it is possible to give a simple and unambiguous description of the topological connectivity of those strands in the sheet (Richardson, 1976, 1977). Each connection between two β strands must fall into one of two basic categories: hairpin connections in which the backbone chain reenters the same end of the β sheet it left, and "crossover" connections in which the chain loops around to reenter the sheet on the opposite end (see Fig. 24). Each connection is named according to how many strands it moves over in the sheet and in which direction, with an "x" added for crossover connections. Thus, a "+1" is a hairpin and a "+1x" a crossover connection between nearest-neighbor strands; a "+2" is a hairpin and a "+2x" is a crossover connection that skips past one intervening strand in the sheet, and so on. The conformation of the connecting loop is irrelevant to this topological designation. Nearest-neighbor connections of +1 and +1x are by far the most common, occurring about three times as frequently as all other connection types put together (Richardson, (1977) Sternberg and Thornton, 1976).
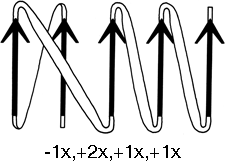
The topology of an n-stranded β sheet can be specified by a list of its n-1 connections, starting from the N-terminus. For example, flavodoxin (Fig. 23) can be described as either +1x, -2x, -1x, -1x, or -1x, +2x, +1x, +1x (absolute value of the signs is not meaningful, since the sheet could be turned upside down). We will use connection types to describe and classify β sheets, and will also use a simplified kind of topology diagram (see Fig. 25) which views the sheet from above. There is another type of topology diagram also common in the literature which views the sheet end-on (see Levitt and Chothia, 1976); the topology is less explicit but more features of the three dimensional structure are retained. That is a significant advantage in the cases in which it works best, but since adherence to the convention forces substantial distortions in some proteins, we will use separate diagrams for the three-dimensional structure and for the topology in the overall survey (see Sections III,A-E).
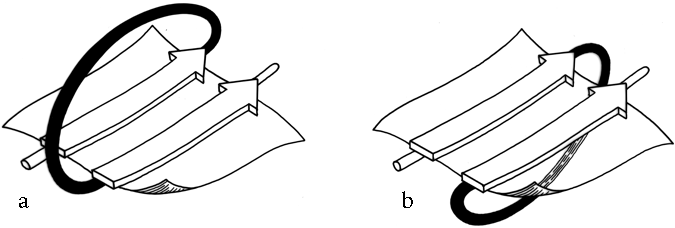
Crossover connections have a handedness (see Fig. 26), since they form a loose helical turn from one strand, up (or down) and around, and back into the next strand. Essentially every one of the crossover connections in the known protein structures regardless of the length or conformation of the connecting loop, is right-handed (Richardson, 1976; Sternberg and Thornton, 1977a). There is one really well authenticated left-handed crossover in subtilisin and one in glucose-phosphate isomerase in a region where the chain connectivity is not completely certain (Shaw and Muirhead, 1977), while there are many more than a hundred right-handed crossovers. [The enormous preference for right- over left-handed crossover connections has held true.]
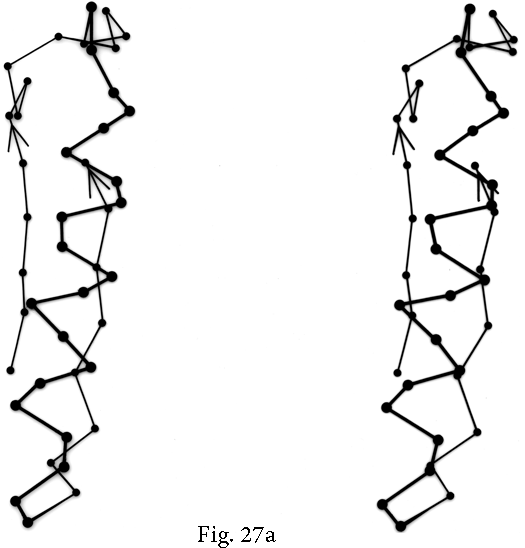

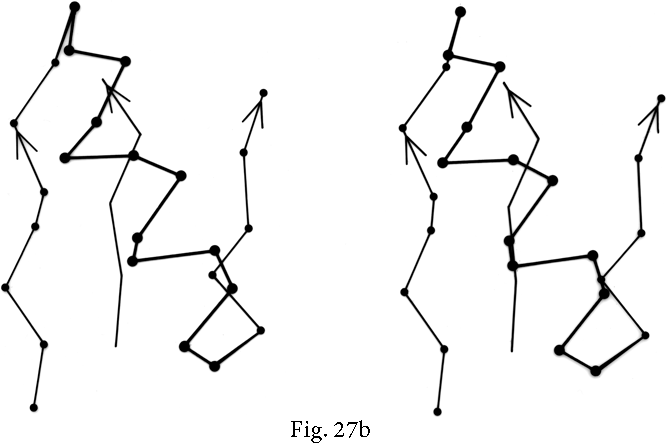
Over half of the crossover connections have at least one helix in the connecting strand, and in many of those cases the helix packs against one or both of the β strands it connects (see Fig. 27a). Sternberg and Thornton (1976) have explained the handedness by the fact that β sheet twist makes the right-handed connection shorter and more compact (as can be seen in Fig. 26). Nagano (1977a) has explained the handedness by the preferred packing angles of a helix against a β strand, which again would allow more compact and shorter corners (between the α and β elements) in the right-handed form. Both of these explanations are sure to be important contributing causes of crossover handedness, but they are limited to the relatively short, straightforward examples with tight corners. The large number of crossover connections which are too long, start off in the wrong direction, or do not pack against the β sheet (see Fig. 27a for examples) show almost as strong a handedness constraint as the more classic cases. In an attempt to account for these long examples, Richardson (1976) proposed a hypothetical folding scheme for crossover connections by which the twist of a long extended strand or of a helix flanked by extended chains is transferred to the crossover loop as the backbone curls up (see Fig. 28). However it is achieved, the right-handedness of crossover connections is the dominant factor controlling the appearance of both singly-wound and doubly-wound parallel α/β structures (see Section III,C). Crossover connections are also fairly common in antiparallel β sheet.
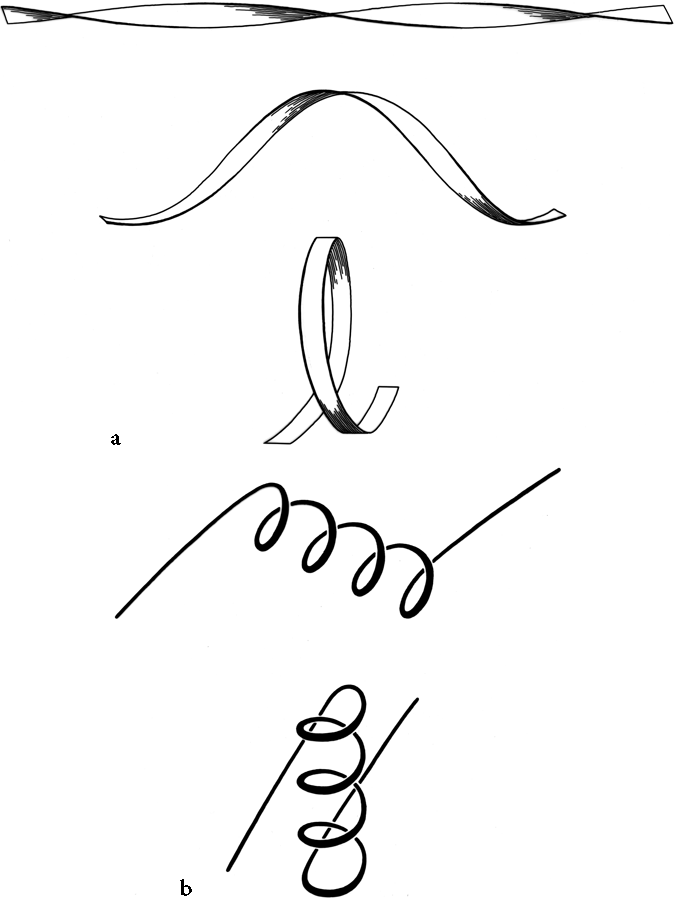
Parallel β structure usually forms large, moderately twisted sheets such as in Fig. 23, although occasionally it rolls up into a cylinder with helices around the outside (e.g., triosephosphate isomerase). Large antiparallel sheets, on the other hand, usually roll up either partially (as in the first domain of thermolysin or in ribonuclease) or completely around to join edges into a cylinder or "barrel." Occurrence, topology, and classification of β barrels will be discussed in Section III,D, but here we will consider the interaction between the β sheets on opposite sides of the barrel, especially in terms of the angle at which opposite strands cross.
β barrels may be made up of as few as 5 or as many as 13 strands. [Even larger, and often quite round, β-barrels occur in membrane-spanning proteins such as the porins, but their insides are not filled by the β-sheet side chains: they are either open or contain loops.] Their interiors are packed with hydrophobic side chains, which are found to have the same average side chain volume as for a normal amino acid composition. There are no large barrels filled with tryptophans or small ones filled with alanines, presumably because mutation to change the size of even as many as two or three residues at once would still produce a bad fit. The cross sections of all the barrels look remarkably alike, regardless of strand number, with a slight flattening in one direction. Figure 29 shows examples of cross sections from real β barrels with different numbers of strands. The nearly constant appearance is obtained by varying the degree of strand twist around the barrel.
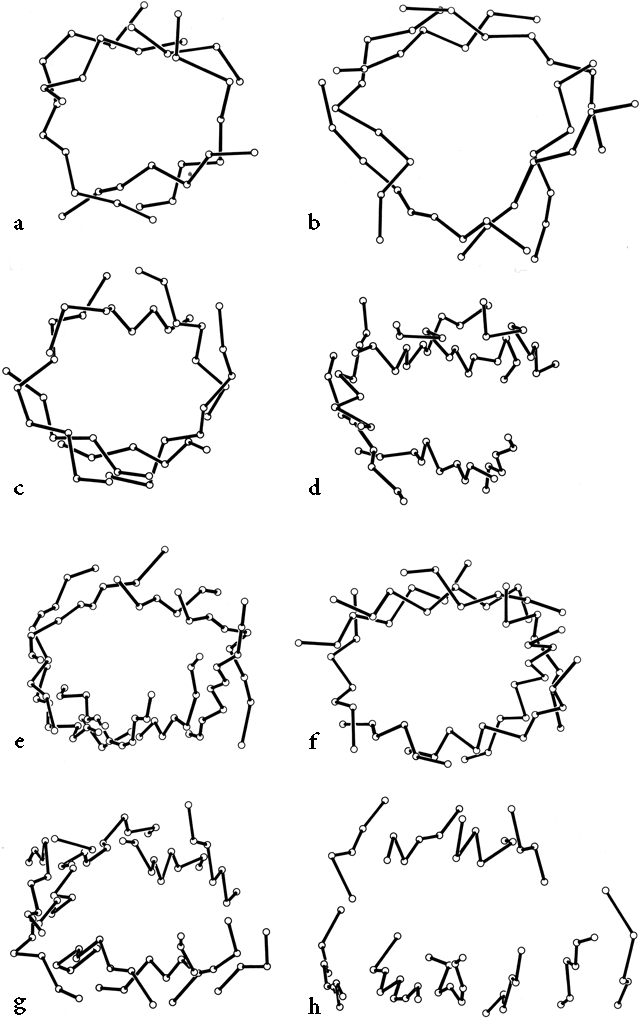
Like the bias-woven finger-bandages which tighten around a finger when stretched, a barrel with a given number of strands has a smaller diameter the less twist it has. Twist can be measured by the angle at which strands on opposite sides of the barrel cross one another; that angle averages 95° for 5- and 6-stranded antiparallel barrels, 40° for 7- and 8-stranded ones, and 30° for 9- through 13-stranded ones. Barrel diameter can also be maintained with fewer strands by separating one or more strand pairs further apart than normal hydrogen-bonding distance; this is a very pronounced effect in plastocyanin, for instance, which has a very low twist angle for an 8-stranded barrel. Eight-stranded parallel barrels are more twisted (averaging 75°) than 8-stranded antiparallel ones because all of their strands are hydrogen-bonded and more regular. Beyond eight or nine strands the twist cannot decrease any further and the barrel cross section simply flattens more, keeping the same short axis (11-12Å).
Tight turns (also known as reverse turns, β turns, β bends, hairpin bends, 310 bends, kinks, widgets, etc.) are the first and most prevalent type of nonrepetitive structure that has been recognized. While helices and β structure have the property that approximately the same φ,ψ angles are repeated for successive residues, pieces of nonrepetitive structure have a particular succession of different φ,ψ values for each residue, so that the concept of residue position within the structure is more influential than in a repeating structure. Of course, no startlingly new local conformations are available: most residues are either approximately α type or β type, with occasional left-handed α-type residues which are usually but not always glycines. However, by combining those three basic conformations in various orders, allowing for the considerable variation available within each of the conformational minima, and utilizing various patterns of hydrogen bonding and side chain position, an enormous number of quite different structures are possible even within a stretch as short as three or four residues.
Tight turns were first recognized from a theoretical conformational analysis by Venkatachalam (1968). He considered what conformations were available to a system of three linked peptide units (or four successive residues) that could be stabilized by a backbone hydrogen bond between the CO of residue n and the NH of residue n + 3.
He found three general types, one of which (type III) actually has repeating φ,ψ values of -60°, -30° and is identical with the 310-helix. The other two types are nonhelical and fold the chain back on itself around a rather square corner so that the first and fourth α-carbons are only about 5 Å apart, as seen in Fig. 30. The backbone at either end of type I or II turns is in approximately the right position to continue in an antiparallel β ribbon. Type I turns have approximately φ2 = -60°, ψ2 = -30°, φ3 = -90°, ψ3 = 0°, and type II approximately φ2 = -60°, ψ2 = 120°, φ3 = +90°, ψ3 = 0°; these two types are related to one another by a 180° flip of the central peptide unit. Types I and III are identical for residue 2 and differ by only 30° in φ3 and ψ3 (compare Fig. 31a).
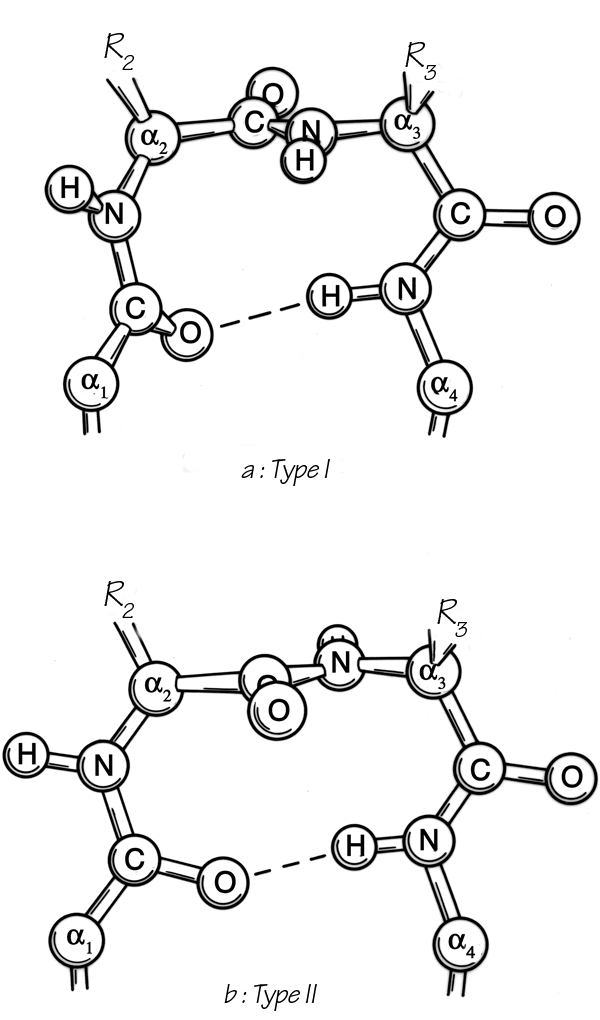
Types I′ and II′ (see Figs. 31 and 32) are the mirror images (for backbone conformation) of types I and II, with the inverse φ,ψ values of those given above. For types II, II′, and I′ the dihedral angles are such that for one or both of the central positions glycine is strongly preferred. In the rather common type II turn, for instance, the carbonyl oxygen of the middle peptide is too close to the β-carbon of the side chain in position 3 (see Fig. 30b), but the bump is relieved if residue 3 is glycine. For type II′ the bump is between Cβ of residue 2 and the NH of the middle peptide. A survey by Chou and Fasman (1977) that identified and characterized 459 tight turns in actual protein structures found that 61% of the type II turns had a glycine in position 3. Type II′ turns strongly prefer glycine in position 2; types I′ and III′ prefer glycine in position 2, but in the actual cases observed seem to adjust conformation slightly rather than have glycine in position 3.
[Sibanda and Thornton (1985) noticed that tight turns in the classic position joining a β hairpin very strongly preferred the mirror-image I′ and II′ conformations. The otherwise-commonest Type I turn has a very pronounced lefthanded twist (diagnosed by a +45° dihedral angle of the 4 turn Cα's), which is in conflict with the righthanded twist of β strands, while the -45° dihedral of Type I′ matches the twist of a β hairpin almost perfectly. Tight turns joining the strands of a β hairpin, therefore, usually use one or two glycines.]
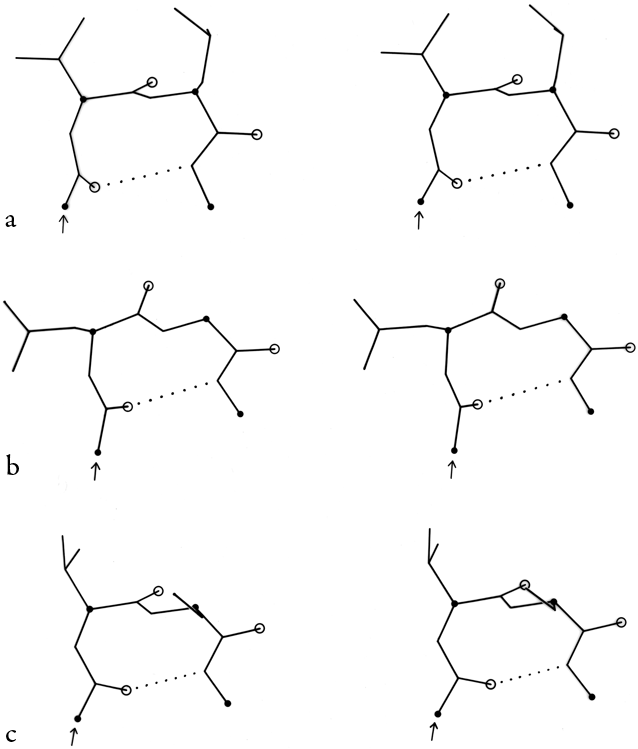
In addition to the above three turn types and their mirror images, Lewis et al. (1973) defined five additional types which both they and Chou and Fasman (1977) find can account for all observed cases (outside of helix) where the α-carbons of residues n and n + 3 are less than 7 Å apart. Type V is a rather unusual departure from type II which has φ2 = -80°, ψ2 = +80°, φ3 = +80°, ψ3 = -80°, and type V′ is its mirror image. Type VI has a cis-proline in position 3; the cis-proline turn was very elegantly demonstrated by Huber and Steigemann (1974) in the refinement of the Bence-Jones protein REI (see Fig. 33b). Type VII has either φ3 near 180° and ψ2 < 60° or else φ3 < 60° and ψ2 near 180°. Type IV is essentially a miscellaneous category, which includes any example with two of the dihedral angles more than 40° away from ideal values for any of the other types.
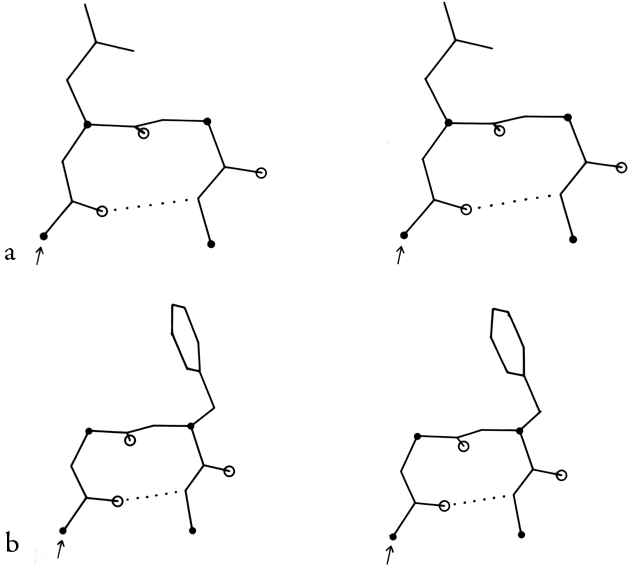

In order to evaluate the occurrence and distinctness of the major turn types as found empirically in protein structures, Figs. 35 through 37 plot φ,ψ values found for turns in Chou and Fasman (1977). Figure 35 shows that types I and III form a single tight cluster even for position 3; their ideal φ,ψ values are so close that they could be distinguished only in the most highly refined protein structures. We would suggest eliminating type III as a distinct category.
The ideal values for an inclusive type I category could either be left as they are and would include essentially all the type III examples, or else ψ3 could be changed to about -10° to be closer to the center of the total cluster of values. There is a rather large number of "nonideal" type I turns that occur at the top in Fig. 35b; it might perhaps be productive to group them as type Ib (since their φ3,ψ3 values are in the β region). Some of these turns have an overall "L" shape (similar to Fig. 34a) and some look like a type I turn with the third peptide flipped over.
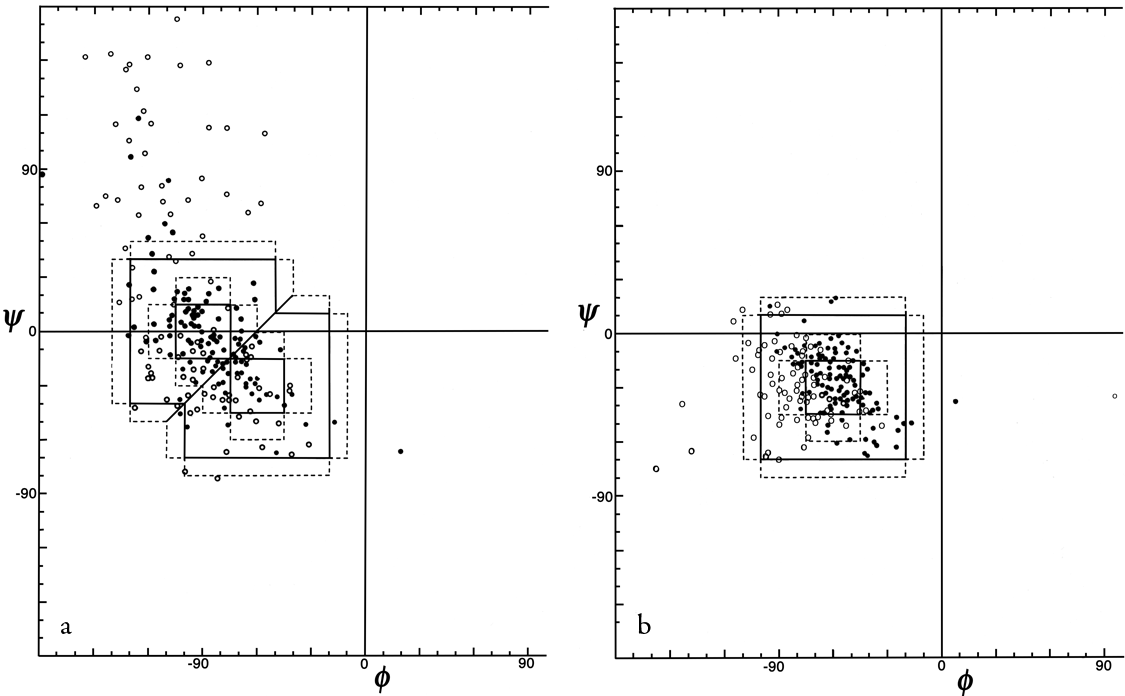
Figure 36 shows good clusters for types II and II′ but no evidence of definable type V or V′ examples, which we would also suggest eliminating as separate categories.
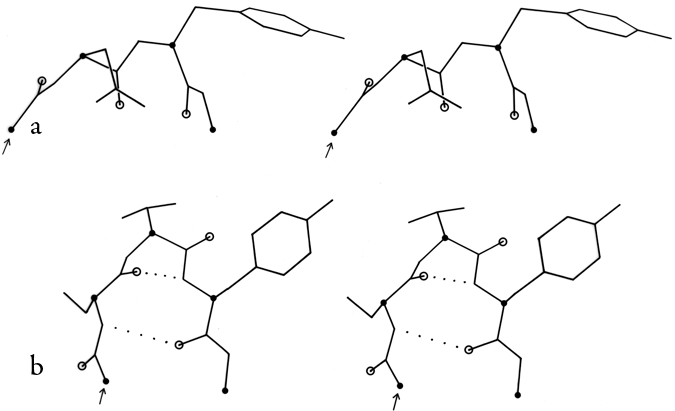
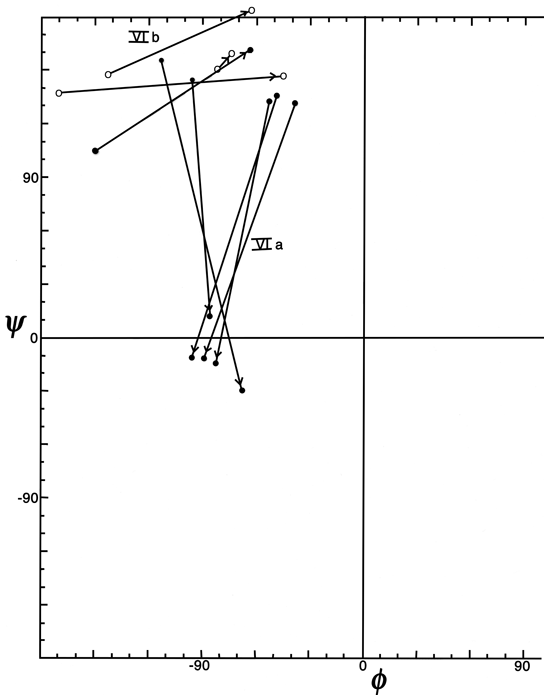
Figure 37 plots φ and ψ for the type VI (cis-proline) turns. Although it is a small sample, there is very strong evidence for two distinct conformations which would be considerably easier to distinguish in an electron density map than to tell cis- from trans-proline in the first place. One of the conformations (which could be called type VIa) has approximately α φ,ψ values for the proline, has a "concave" orientation of the middle peptide and the proline ring relative to the overall curve of the turn (see Fig. 33a), and typically is hydrogen-bonded. The other conformation (which includes the original examples found by Huber and could be called type VIb) has approximately β φ,ψ values for the proline, has a "convex" orientation of the middle peptide and the proline ring (see Fig. 33b) and is usually not hydrogen-bonded.
[An additional, even tighter, turn type involving a cis proline has recently been described in Videau et al. (2004); ref ?? called the cisPro touch-turn, it is apparently somewhat strained and is common only at binding and active sites.]

Since type VII turns are defined by only two angles and can have two different values for those, they vary greatly in appearance. Of the nine observed examples at least half are questionable (for instance, staphylococcal nuclease 47-50 is at the end of a partially disordered loop, and for rubredoxin 46-49 the unconstrained refinement has placed the atoms of the middle peptide out of line just enough so that the ordinary definition of φ and ψ is meaningless). Therefore type VII also seems unjustifiable as a distinct category. Some of the type VII turns (and also some type IVs) fall at the edge of the cluster of φ,ψ values seen for type Ib (see above), and perhaps could be included in that group.
In summary, then, tight turns can be rather well described by a set of categories consisting of types I, I′, II, II′, VIa, VIb, and miscellaneous (IV), with the possible addition of type Ib.
In order to demonstrate what the various types of turns actually look like, Figs. 31 through 34 show stereo views of turn examples from real structures that have φ,ψ angles very close to the defining values for each type. Type III is illustrated for completeness, but it cannot be distinguished from type I by inspection unless it is part of a continuing 310-helix. Types IV and V are not shown, because type IV is a miscellaneous category and there are no ideal cases of type V (see Fig. 36). The turns are all shown in approximately the same standard orientation: with the mean plane of the four α-carbons in the plane of the page and the chain entering at the lower left. In this orientation the β-carbons are always at or above the plane for types I, I′, II, and II′ turns (since only the backbone conformation can be mirrored). The virtual bond dihedral angle defined by the four α-carbons is close to 0° for type II or II′ and is somewhat positive (averaging about +45°) for type I and somewhat negative for type I′. All four types have the third peptide essentially in the mean α-carbon plane. If the carbonyl oxygens are visible in an electron density map, then these four turn types can be fairly readily distinguished. In types I and II′ the second carbonyl oxygen points approximately 90° down from the plane, while in types II and I′ it points approximately 90° up. The first oxygen points nearly 90° down from the center of the plane in type I, nearly 90° up in type I′, slightly up [ down ] in type II, and slightly down [ up ] in type II′. The position of the second carbonyl oxygen, then, distinguishes between types I and II (or I′ and II′), while the position of the first carbonyl oxygen distinguishes types I vs II′ (or II vs I′). For either distinction intermediate cases should be rare, because they lie in a strongly prohibited region of the φ,ψ map.
The simple conception of a tight turn as approximately planar with a linear hydrogen bond is fairly accurate for type II. However, even an "ideal" type I turn is decidedly nonplanar, with the NH and CO of its hydrogen bond almost perpendicular to each other. That oxygen is in the plane of the last three α-carbons, but the first α-carbon and peptide are swung up out of the plane, producing the 45° virtual dihedral angle.
One additional sort of tight turn involving only three residues has been described theoretically (Némethy and Printz, 1972) and also observed at least once in a protein structure (Matthews, 1972). This is the γ turn, which has a very tight hydrogen bond across a seven-atom ring between the CO of the first residue and the NH of the third (see Fig. 34b). It also can continue with a normal β sheet hydrogen bond between the NH of residue 1 and the CO of residue 3. Residues 1 and 3 are not far from the usual β conformation, while φ2 = 70° and ψ2 = -60°. [The γ turn is real but rare, having a somewhat distorted H-bond and bond angles spread slightly to avoid a clash between Cβ(2) and CO(1). It has been treated in surprisingly disparate ways by different sections of the structural biology community. It is invoked rather too routinely in peptide nmr studies, perhaps influenced by the fact that the γ-turn φ,ψ values (and the mirror-image γ′ at =70°,60°) are the overwhelming global minima for energy calculations in vacuo (e.g. Head-Gordon, 1991), while the γ-turn conformation is avoided when possible by protein crystallographers because that region of the φ,ψ plot is classed as forbidden by Procheck (Laskowski, 1993), the standard structure-validation program. We would recommend an intermediate level of enthusiasm (Lovell, 2003).]
Although the presence of the hydrogen bond led to the initial characterization of tight turns by Venkatachalam (1968), hydrogen bonding was dropped as a necessary condition as soon as any surveys were done on known protein structures (e.g., Crawford et al., 1973), because numerous examples were found outside plausible hydrogen-bonding distance but with otherwise very turnlike conformation. About half of the turns listed in Chou and Fasman (1977) are hydrogen-bonded (by the criterion that O1 to N4 is less than 3.5 Å). Apparently the various turn conformations are sufficiently favorable so that they do not require stabilization by the hydrogen bond. This should not be surprising, since the turn types essentially consist of the basic α, β, and left-handed glycine conformations in various combinations. Also, since turns typically occur at the surface, peptides can hydrogen bond to solvent when not bonded to each other.
There are a number of characteristic residue preferences for tight turns. The most general is a strong tendency for turn residues to be hydrophilic (e.g., Kuntz, 1972; Rose, 1978), which might reflect inherent conformational preferences but is more probably a result of the almost universal location of turns at the protein surface where they interrupt or join together segments of secondary structure that are more internal. Glycines are quite common in tight turns, as can be inferred from their preference in types II, I′, and II′; however, glycine is actually not quite as common as would be implied by energy calculations (see Chou and Fasman, 1977). Proline is also common in turns; besides the cis-proline turn, proline also fits well in position 2 of types I, II, and III turns and position 3 of type II′. About two-thirds of the Pro-Asn and Pro-Gly sequences in the known protein structures are found as the middle two residues of a tight turn (Zimmerman and Scheraga, 1977b).
Tight turns can combine with other types of structure in a number of ways. In addition to their classic role of joining β strands, they often occur at the ends of α-helices (see Section II,A). A type II turn forms a rather common combination next to a G1 β bulge (see Section II,D). Isogai et al. (1980) have surveyed the occurrence of successive tight turns, which either form approximately helical features or else form more complex chain reversals than single turns.
[As noted above, the most important later insight about tight turns was the demonstration by Sibanda and Thornton (1985) that the classic role at the end of a β-hairpin can be filled well only by the otherwise-rare types I′ and II′, since the virtual dihedral angle formed by types I or II has the wrong handedness to mesh with the right-handed twist of the β-strands in the hairpin. A more recent survey of tight turns can be found in (Wilmot and Thornton, 1988).]
In addition to the approach described at length above, a number of authors have adopted a very useful but much looser approach to defining turns (see Kuntz, 1972; Levitt and Greer, 1977; Rose and Seltzer, 1977). Instead of a detailed conformational analysis in terms of conformational angles, hydrogen bonds, etc., these authors want a concept of turns that can be defined systematically and reproducibly from preliminary α-carbon coordinates and that (except for Levitt and Greer) is meant to include also the larger and more open direction changes in the polypeptide chain. Kuntz looks at direction changes for Cαn—Cαn+1 vectors versus Cαn+x—Cαn+x+1 where x is 2 (for usual tight turns) or more; turns have direction changes greater than 90° and short Cαn to Cαn+x distances. Levitt and Greer assign turns to nonhelical, non-β segments for which the virtual dihedral angle defined by four successive α-carbons is between -90° and +90°. Rose and Seltzer define turns as local minima in the radius of curvature calculated from points Cαn-2, Cαn, and Cαn+2, with the modification that turns correspond only to places where the chain in both directions cannot be fitted inside a single rigid cylinder of 5.2Å diameter. Of course, the conformational approach of Lewis et al. (1973) defining detailed turn types must start with a general definition also, which happens to be a Cαn to Cαn+3 distance less than 7Å. Conversely, any of the above general definitions could be used as a starting point from which to examine detailed conformations.
Both of these approaches have their strengths and their limitations. A great many people have deplored the fact that there is not very close agreement between the sets of turns (or of any other structure type) identified by any two different people (or computer programs) for a given protein. Each author suggests that the problem can be solved if everyone else accepts his definitions and criteria. Not only does this seem unlikely to happen, but it would probably be very undesirable. The major reason for the discrepancies is that each author has a different set of purposes for which he wants to use the structural characterization, and each is working from a data base of structures known to widely varying degrees of detail and reliability. Even the standard IUPAC-IUB conventions (1970) recognize two different definitions of α-helix, for instance: one based on φ,ψ values and the other on hydrogen-bonding.
The looser sort of turn definition derived from Cα positions might be the most useful one for studying protein folding and perhaps even for predicting turns. It can be applied to the largest available data base since it requires only α-carbon coordinates. However, in a very real sense it is not appropriate by itself for the accurately determined structures because it is incapable of taking into account the enormous amount of additional information they contain. For example, Levitt and Greer calculate possible hydrogen bonds from just Cα positions, which is a useful way of extending partial information; however, where atomic coordinates are available it turns out that a small but significant fraction of those bonds are definitely not present. Some form of detailed, conformational definition for turns is clearly needed for purposes such as energy calculations, examination of side chain influences, or correlation with spectroscopic observations such as CD or Raman. The detailed definitions will probably be increasingly fruitful as the number of highly refined protein structures increases; one severe problem at present is that they have usually been applied uncritically to all available sets of complete atomic coordinates, regardless of the fact that very few of the structures are known well enough for the distinction of 30° in dihedral angle between types I and III to have much meaning, and that in places where carbonyl oxygens were not visible in the electron density map even the distinction between types I and II is ambiguous. The best solution is probably to continue using whatever turn criteria are most appropriate for a given purpose but to state the criteria explicitly and to give careful consideration to selection of a data base. [These days such classification of structure motifs should always use quality filters based on both resolution and B-factor.]
By any sort of definition, turns are an important feature of protein structure. Kuntz (1972) found 45% of protein backbone in turns or loops; Chou and Fasman (1977) found 32% of protein chain in turns (counting four residues per turn); and Zimmerman and Scheraga (1977b) found 24% of the nonhelical residues in turns (counting only the central dipeptide). There are also some particular proteins whose structure appears heavily dependent on turns: Fig. 38 shows high-potential iron protein (Carter et al., 1974), with the 17 turns in 85 residues indicated and their location at the surface evident.
Large portions of most protein structures can be described as stretches of secondary structure (helices or β strands) joined by turns, which provide direction change and offset between sequence adjacent pieces of secondary structure. Tight turns work well as α-α and α-β joints, but their neatest application is at a hairpin connection between adjacent antiparallel β strands, where the hydrogen bond of the turn is also one of the β sheet hydrogen bonds, as in Fig. 39. [But see comment above.]
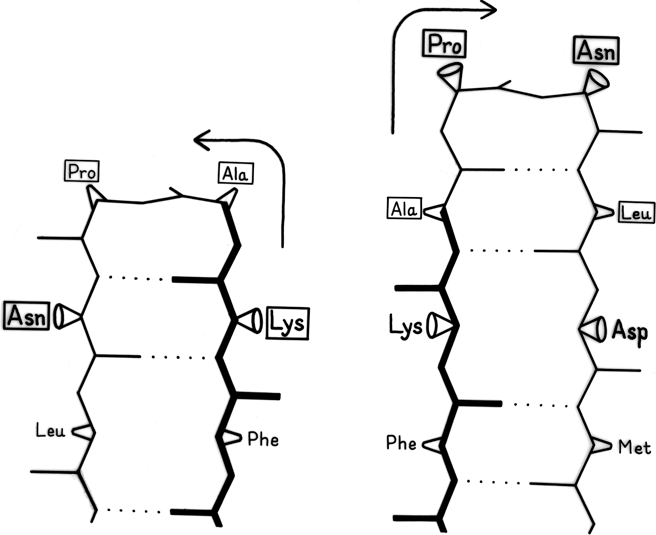
It has often been suggested (e.g., Lewis et al., 1971; Rose et al., 1976) that turns can provide a decisive influence in directing the process of protein folding to the native conformation, since they seem ideally suited to help specify as well as encourage the decisive long-range interactions that form tertiary structure. As one simple example of the way in which turns can direct and specify other interactions, let us consider what happens when a tight turn immediately joins two adjacent antiparallel strands, as shown in Fig. 39. During the folding process it must be determined whether strand B will lie to the left or to the right of strand A. If the position of the tight turn is shifted by one residue along the sequence, then the turn must be made in the opposite direction to preserve hydrogen-bonding and amino acid handedness, as shown in the two parts of the figure. If the first strand runs from bottom to top and if the side chain in position 1 of the tight turn points toward you, then the second strand must lie to the right; if the side chain in position 1 of the turn points away, then the second strand must lie to the left. Since there are strong positional preferences for the residues in tight turns, it seems plausible that they can actually exert this sort of influence on the folding of neighboring strands.
The β bulge (Richardson et al., 1978) is a small piece of nonrepetitive structure that can occur by itself in the coil regions, but which most often occurs, and is most easily visualized, as an irregularity in antiparallel β structure.
A β bulge is defined as a region between two consecutive β-type hydrogen bonds that includes two residues on one strand opposite a single residue on the other strand. Figure 40 shows a β bulge from trypsin. The two residues on the bulged strand are called positions 1 and 2, and the one on the opposite strand position X. Sometimes the hydrogen bond to the CO of position X is forked, coming from the NH groups of both positions 1 and 2; the bulge in Fig. 40 shows this feature.
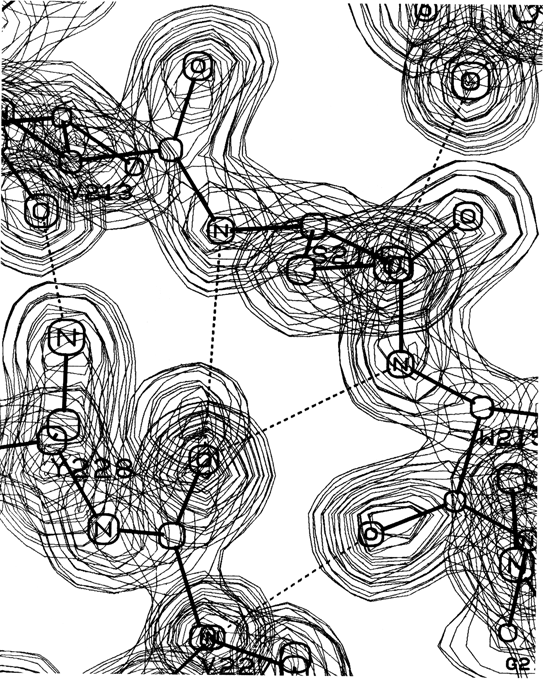
Only about 5% of the β bulges are between parallel strands, and most of the antiparallel ones are between a closely spaced (see Section II,B) rather than a widely spaced pair of hydrogen bonds. The additional backbone length of the extra residue on the longer side is accommodated partly by bulging that strand to the right and toward you as seen in Fig. 40artly by putting a slight bend in the β sheet.
Like tight turns, bulges affect the directionality of the polypeptide chain, but in a much less drastic manner. β bulges are not as common as tight turns, but over a hundred examples are known (see Richardson et al., 1978, for a listing of 91). [A survey by Chan et al. (1993) covers much larger numbers.]
β bulges can be classified into several different types, which are illustrated schematically in Fig. 41. By far the commonest is the "classic" β bulge, which occurs between a narrow pair of hydrogen bonds on antiparallel strands and has the side chains of positions 1, 2, and X all on the same side of the β sheet (see Fig. 41a). Residue 1 is in approximately α-helical conformation (averaging φ1 = -100°, ψ1 = -45°) and residues 2 and X in approximately normal β conformation (averaging φ2 = -140°, ψ2 = 160°, and φx = -100°, ψx = 130°). Figure 42 is a stereo drawing of five examples of classic β bulges superimposed on one another. Note that the carbonyl oxygens on either side of residue 1 both point in about the same direction, as is typical of α-helical conformation, while the carbonyls surrounding residues 2 and X point opposite each other, as in β structure. Figure 42 also shows that a classic bulge locally accentuates the normal right-handed twist (see Section II,B) of the β strands.
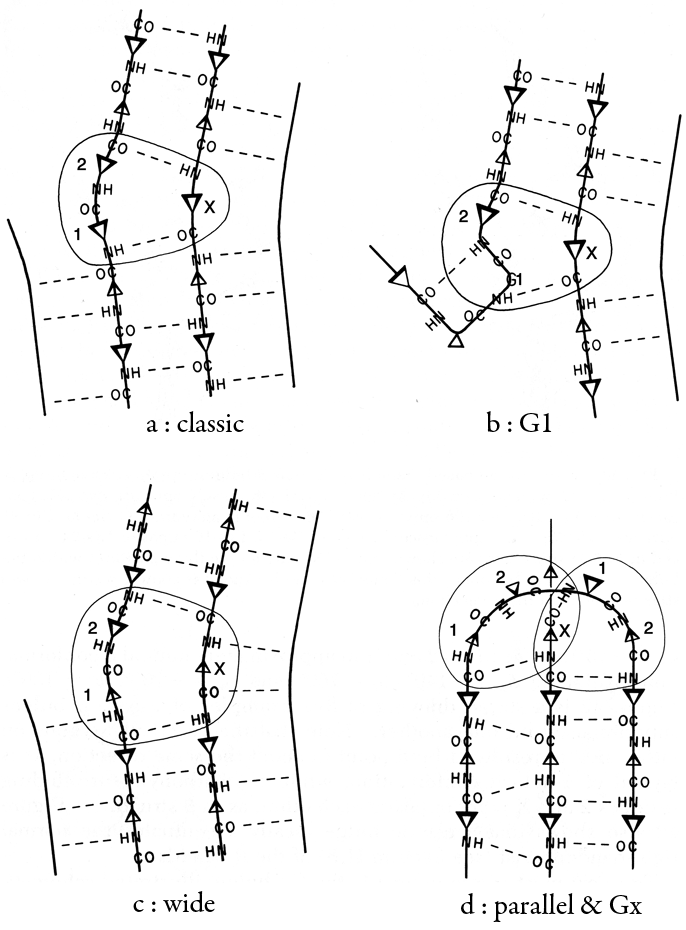
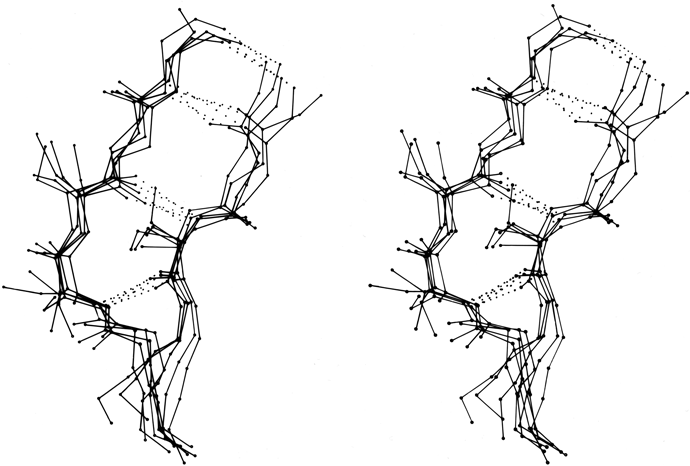
The next most common type is the G1 bulge, illustrated schematically in Fig. 41b. It also lies between a narrow pair of hydrogen bonds, and position 1 is almost invariably a glycine because of its backbone conformation: φ1 ≅ 85°, ψ1 ≅ 0° and φ2 ≅ -90°, ψ2 ≅ 150°. More than half of the G1 bulges are found within an interlocking structure in which the glycine in position 1 of the G1 bulge is also the required glycine in position 3 of a type II tight turn (see preceding section). The plane of the tight turn and its hydrogen bond is almost perpendicular to the plane of the G1 bulge. This combined structure has a consistent handedness which is dictated by the requirements of the three hydrogen bonds. A set of G1 bulges with their associated tight turns are shown superimposed in stereo in Fig. 43. For G1 bulges, the side chain of the glycine (if it had one) would be on the opposite side of the β sheet from those in positions 2 and X. In contrast to classic bulges, G1 bulges seldom have continued β structure on the bottom end (as seen in Fig. 41b or 43); when there is an associated tight turn the bulged strand enters at a very sharp angle, and in many of the remaining cases there is a short connection between the two strands of the bulge (so that position 1 equals either χ + 3 or χ + 4). [Such "β-bulge loops" are treated separately by Milner-White (1987).]

"Wide type" β bulges are those that occur between a widely spaced pair of hydrogen bonds on antiparallel strands, as shown schematically in Fig. 41c. They apparently are much less constrained than narrow bulges, since they occur with a great variety of backbone conformations; however, they do not occur as often. Even more unusual are the Gx bulges (so named because they often have a glycine in position X) with a hydrogen-bonding pattern similar to that shown in Fig. 41d, and the parallel bulges, which can take several forms, one of which is also illustrated in Fig. 41d.
Bulges have the general property of causing the normal β sheet alternation of side chain direction to be out of register on the two ends of one of the bulge strands. The bulge can be thought of as turning over one-half of the out-of-register strand; the classic bulge accomplishes this by changing the ψ angle of the residue in position 1 by about 180°, which moves it from the β conformational region to the α region. Once half of the strand has flipped over, it must shift sideways by one residue along the other strand in order to hydrogen bond; that shift produces the bulge of two residues opposite one.
Bulges, as well as tight turns, are very often found at active sites, probably because they have a strictly local but specific and controlled effect on side chain direction. Another possible function for β bulges would be as a mechanism for accommodating a single-residue insertion or deletion mutation without totally disrupting the β sheet. There seem to be several such cases in the immunoglobulins, the clearest of which is a one-residue insertion in the CH1 domain of Fab'NEW relative to the sequence of McPC603 CH1: the NEW structure has a bulge in the middle of a long pair of β strands, while in McPC603 those strands form regular β structure all the way along.
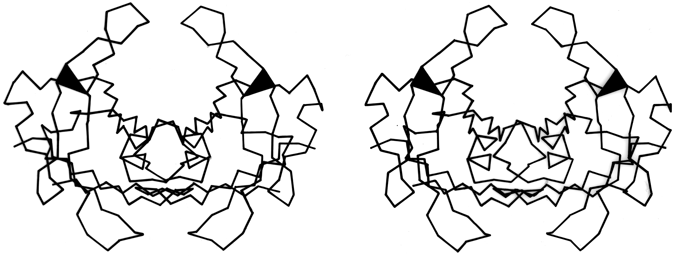
The other general property of β bulges is that they alter the direction of the backbone strands forming them; classic bulges also accentuate the right-handed twist of the strands. This means that bulges are often useful for shaping large features of β sheet and/or extended hairpin loops. In antiparallel β barrels, for instance (see Section II,B), an extremely strong local twist is needed for closing barrels as small as five or six strands. Bulges in chymotrypsin, trypsin, elastase, staphylococcal nuclease, papain domain 2, and probably soybean trypsin inhibitor are strategically located at the sharpest corners in the β strands. Extended two-strand β ribbons are often used for forming external interaction sites on a protein. Such a ribbon would normally extend from the end of a β sheet or β barrel, but if the interaction site needs to be at one side of the sheet or barrel, then a β bulge at the point of departure can be used to direct the β ribbon out more nearly at right angles. For example, in the immunoglobulins the 47,48;35 bulge in the VL domain and the 48,49;36 bulge in the VH domain send out a pair of β ribbons that help complete closure around the VL—VH interdomain contact. In prealbumin the Phe-44,Ala-45;Val-32 bulge helps to turn out the extended VL ribbon near the 2-fold axis of the dimer that forms the DNA-binding site proposed in Blake and Oatley (1977) (see Fig. 44).
Disulfide bridges are, of course, true covalent bonds (between the sulfurs of two cysteine side chains) and are thus considered part of the primary structure of a protein by most definitions. Experimentally they also belong there, since they can be determined as part of, or an extension of, an amino acid sequence determination. [This was true when sequencing was done on proteins, but of course DNA sequencing does not determine SS connectivity.] However, proteins normally can fold up correctly without or before disulfide formation, and those SS links appear to influence the structure more in the manner of secondary-structural elements, by providing local specificity and stabilization. Therefore, it seems appropriate to consider them here along with the other basic elements making up three-dimensional protein structure.

A modest amount of accurate conformational information is available from small-molecule X-ray structures of various forms of cystine. χ1 angles are close to +60°, and all three dihedral angles internal to the disulfide are close to + or -90°. Two mirror-image conformations are observed; in N,N'-diglycyl-L-cystine dihydrate (Yakel and Hughes, 1954) and in L-cystine dihydrobromide (Peterson et al., 1960) or hydrochloride (Steinrauf et al., 1958) all three internal dihedral angles are approximately -90°, forming a left-handed spiral, while in hexagonal L-cystine (Oughton and Harrison, 1957, 1959) they are all approximately +90°, forming a right-handed spiral. Figure 45 shows the left-handed disulfide of L-cystine dihydrobromide, viewed down the 2-fold axis perpendicular to the S—S bond.
The dihedral angles of disulfides in proteins are very difficult to determine with any accuracy except in refined high-resolution structures. In the first few protein structures to show disulfides at 2 Å resolution, attention was paid mostly to the dihedral angle around the S—S bond (χ1), since it is the most characteristically interesting parameter for cystine, is one of the easier ones to measure, and presumably is correlated with the handedness of the large optical rotation associated with the presence of disulfide bridges. As expected, the χ3 angles were in the range around 90-100° and were found in both left-handed and right-handed forms (Blake et al., 1967; Wyckoff et al., 1970).
[By now of course, a great many more SS-containing structures have been determined, and better statistics are available (see below). Oddly, however, SS conformation is the least well determined of any side chain because 1) they are relatively rare, 2) series-termination ripples around the S atoms can distort the electron density, and 3) the fact that they are connected on both ends makes any misfittings difficult to correct.]
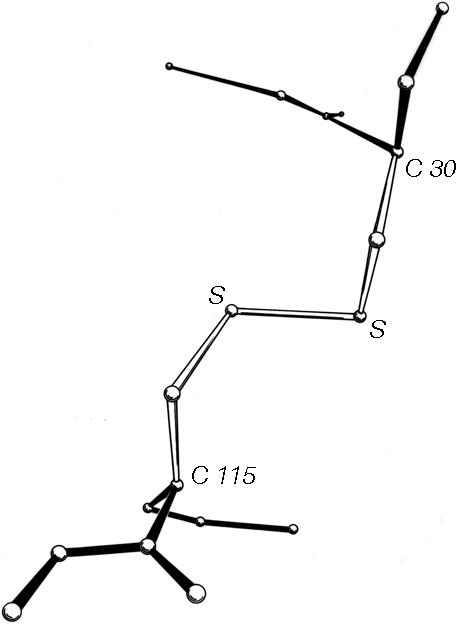
Now that about 70 different disulfides have been seen in proteins and more than 20 of those have been refined at high resolution, it is possible to examine disulfide conformation in more detail, as it occurs in proteins. Many examples resemble the left-handed small-molecule structures extremely closely; Fig. 46 shows the Cys-30-Cys-115 disulfide from egg white lysozyme. The χ2, χ3, and χ2' dihedral angles and the Cα—Cα' distance can be almost exactly superimposed on Fig. 45; the only major difference is in χ1. All of the small-molecule structures have χ1 close to 60°. Figure 47 shows the χ1 values for half-cystines found in proteins. The preferred value is -60° (which puts Sγ trans- to the peptide carbonyl), while 60° is quite rare since it produces unfavorable bumps between Sγ and the main chain except with a few specific combinations of χ2 value and backbone conformation.

[Add Fig. for current data ??]
If χ3 is kept between 90° and 100° and χ2 and χ2' are varied, the distance between α-carbons across the disulfide can range all the way from about 4 to 9Å. That corresponds to the extreme range observed for all disulfides. However, the effective range for Cα—Cα' distance is really a good deal narrower than that, since 85% of all the disulfides and 95% of the refined ones fall between 4.4 and 6.8Å.
Now let us examine the relationships between handedness of χ3, Cα—Cα distance, and χ2 values. Among the disulfides for which coordinates were available at 2 Å resolution or better (Deisenhofer and Steigemann, 1975; Imoto et al., 1972; Wyckoff et al., 1970; Quiocho and Lipscomb, 1971; Saul et al., 1978; Epp et al., 1975; Huber et al., 1974; Chambers and Stroud, 1979; Hendrickson and Teeter, 1981; Brookhaven Data Bank, (1980) Feldmann, 1977), there are equal numbers with right-handed and left-handed χ3. The average Cα—Cα' distance across the left-handed ones is 6.1 Å, exactly what was seen in the small-molecule structures, but for the righthanded ones the average Cα—Cα' distance is 5.2 Å. [The equal numbers and the length difference, still holds true.] Clearly the two sets of disulfides as they occur in proteins cannot simply be mirror images of one another.
Figure 48 plots all five side chain dihedral angles (χ1, χ2, χ3, χ2', and χ1') and the Cα—Cα' distances for the high-resolution disulfides. The overall conformations fall into just two major and two minor categories (plus perhaps one more to be discussed below). Essentially all of the left-handed ones (solid lines in Fig. 48) have approximately χ1 = -60°, χ2 = -90°, χ3 = -90°, χ2' = -90°, χ1' = -60°. This can be called the left-handed spiral conformation, and is the same as that seen in Fig. 46. An example from ribonuclease S is shown in Fig. 49, looking down the spiral. A majority of the right-handed disulfides have a conformation of approximately χ1 = -60°, χ2 = +120°, χ3 = +90°, χ2' = -50°, χ1' = -60° (heavy dots in Fig. 48) and a Cα separation averaging only 5 Å. This can be called the right-handed hook conformation; a typical example is shown in Fig. 50. Two cases have - + - dihedral angles and a Cα—Cα distance of 4-4.5 Å; these could be called short right-handed hooks. [Now that the database is larger, it can be seen that these short - + - disulfides join residues opposite one another on adjacent antiparallel β strands. They have been christened "staples" by Sternberg. Although connecting β strands is a very useful function the SS staples are not very common, reflecting the fact that their conformation is actually somewhat strained.] Then there are two cases of right-handed spirals (dashed lines in Fig. 48) which tend to have one χ1 angle of 180° and are still significantly shorter than the left-handed spirals. (ref ??) [?? is this still true?]
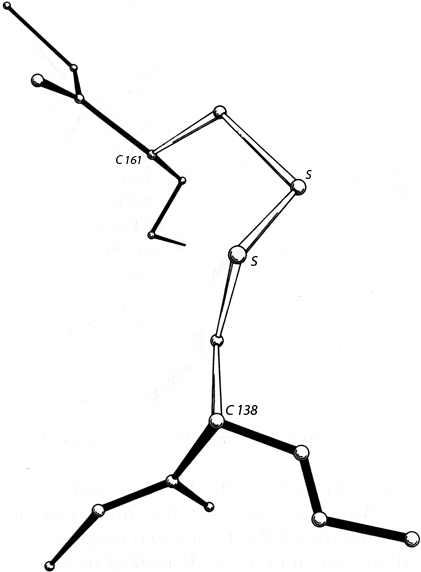
The only cases that were omitted from Fig. 48 are the disulfides that span the β barrels in immunoglobulins (Epp et al., 1975; Saul et al., 1978). They are unusually long, with Cα separations of 6.6 to 7.4 Å, which is achieved by having both χ2 and χ2' close to 180°. These long disulfides are trans-gauche-trans (180°, +or-90°, 180°) in χ2, χ3, χ2', while both the spiral and the hook conformations described above are gauche-gauche-gauche (+or-90°, +or-90°, +or-90°). The χ1 angles are generally either 180 or 60° (in contrast to the usually preferred -60°) and the χ3s show no preference for left-handed vs right-handed. Indeed, in the Bence-Jones VL dimer REI (Epp et al., 1975) the disulfides were found to be a disordered mixture of the right-handed and left-handed forms. Asymmetrical preferences for the handedness of χ3 presumably must involve either direct or indirect interactions with the backbone; with χ2 near 180° such interactions are minimized. However, one must also presume that these unusually long disulfides are at least slightly strained and that χ2 angles in the 60-120° range are more favorable as a general rule. It also may well be true that χ3 is even more strongly constrained to +90° than is χ2, but the angle distributions seen in Fig. 48 should not be taken as proof that that is so, because preferred values for χ3 and not for χ2 have been built into almost all model-building and refinement routines. Molecular orbital calculations for χ3 by Pullman and Pullman (1974) give an energy minimum at 100°, but do not rise by more than 0.5 kcal/mol from about 70 to 140°. They did not report calculations for χ2. [?? Note Hermans or other calc's?]
The distinctive differences between the left- and right-handed disulfide conformations have little to do with the χ3 angle itself. The bumps of the sulfurs with the polypeptide backbone are produced by a combination of χ1 and χ2; since it is unfavorable to have χ2 in the range of +60 to +100° when χ1 has its preferred value near -60°, the right-handed disulfides cannot adopt a +++++ spiral in proteins.
In summary, we can expect that most disulfides will have Cα separations of less than 6.5 Å unless they are stretched across a β barrel or perhaps a short loop. The majority will have either the left-handed spiral conformation or the right-handed hook conformation. [?? Summary for recent data?]
Now let us examine the distribution and position of disulfides in proteins. The simplest consideration is distribution in the sequence (see Fig. 51), which is apparently quite random, except that there must be at least two residues in between connected half-cystines. [Rare cases are now known with tighter spacing, which is thus quite unfavorable but not impossible.] Examples: ??] Even rather conspicuous patterns such as two consecutive half-cystines in separate disulfides turn out, when the distribution is plotted for the solved structures (Fig. 51), to occur at only about the random expected frequency. The sequence distribution of half-cystines is influenced by the statistics of close contacts in the three-dimensional structures, but apparently there are no strong preferences of the cystines that could influence the three-dimensional structure.
Disulfide topology may be considered in terms of the possible patterns of cross-bridges on a floppy string. The cases that occur appear to be a random selection among the possible alternatives, showing no evident preferences for or against any particular features (such as whether nearest-neighbor half-cystines are connected or whether the total connectivity can be drawn in two dimensions). This situation is a very marked contrast to what we found for β sheet topology, where there are a number of quite strong topological preferences. However, the topological and sequence randomness of disulfides is what one would expect if their major role is to stabilize close contacts in the final structure but they have no influence on the early stages of the protein-folding process.
We can also examine what types of backbone conformation are found at the ends of disulfides, and here we can see some preferences again. Well over half of the backbone strands are in extended conformation, although a relatively small fraction are actually part of a β sheet. It is not possible for a disulfide to join neighboring strands in a β sheet [(This has turned out not to be true: SS "staples" do so (see above).)] : any but the closest residues on adjacent strands are too far apart, and a closest pair of residues is slightly too close together. Also, for a close pair on β sheet the Cα-Cβ bonds of the two are approximately parallel, while they need to be approximately perpendicular for a right-handed hook and antiparallel for either right- or left-handed, spirals. Occasionally disulfides join next-nearest-neighbor β strands with only some disruption of the intervening hydrogen-bonding pattern (e.g., Cys-40-Cys-95 in ribonuclease S). An even more common relationship to β sheet is a disulfide joining the continuation of two nearest-neighbor strands after they have separated from the β sheet and can attain a more suitable separation and angle. Typically the disulfide is one or two residues out from the last hydrogen bond on one strand and three or four residues out on the other strand; the β strands are almost always antiparallel rather than parallel. This sort of arrangement is somewhat reminiscent of a β bulge (see Section II,D), and for the case of Cys-65-Cys-72 in ribonuclease S the disulfide actually spans one end of a wide type β bulge. [Another common arrangement in antiparallel β structure has two SS in contact, coming from residues directly opposite on the adjacent strands of a β-hairpin, one SS in a right-hand spiral and one in a left-hand spiral (Richardson Protein Tourist). This has been aptly named the SS β cross (Sternberg).]
α-Helix is also quite common as the backbone conformation flanking a disulfide, but there is seldom well-formed helix on both ends of a given disulfide. If one end comes from an α-helix, the other end will usually be an extended chain, or one or more tight turns, or irregular structure past the end of a helix. Presumably the constraints on favorable separation and angle of Cα—Cβ bonds in disulfides are difficult to satisfy with residues in any of the normal helix packing arrangements (see Section II,A). There are no disulfides at all in any of the helix-bundle structures (see Section III,B). Disulfides do connect a pair of adjacent helices, however, in phospholipase A2 and in crambin.
In cases in which backbone direction is readily definable (primarily for extended or helical chains), the two chains joined by a disulfide almost always cross at steep angles to one another (60 to 90°).
The third most frequent backbone conformation at disulfide ends is a tight turn. Sometimes it is a succession of turns, or bit of 310-helix. Turns seem to be somewhat favored at the hook end of a right-handed hook disulfide.

There is a correlation between the backbone conformations which commonly flank disulfides and the frequency with which disulfides occur in the different types of overall protein structure (see Section III,A for explanation of structure types), although it is unclear which preference is the cause and which the effect. There are very few disulfides in the antiparallel helical bundle proteins and [almost] none in proteins based on pure parallel β sheet (except for active-site disulfides such as in glutathione reductase). Antiparallel β sheet, mixed β sheet, and the miscellaneous α proteins have a half-cystine content of 0-5%. Small proteins with low secondary-structure content often have up to 15-20% half-cystine. Figure 52 shows the structure of insulin, one of the small proteins in which disulfides appear to play a major role in the organization and stability of the overall structure.
When there is a need to divide up protein structure for purposes of description, prediction, spectroscopic characterization, etc., the usual categories have been "helix," "β structure," and "coil." "Coil" is defined in practice as "none of the above." Its major distinguishing feature is that it is nonrepetitive in backbone conformation (although depending on one's definition of β structure, one might or might not include an isolated piece of extended chain as coil). Sometimes coil is referred to as "random coil," or is modeled by properties observed in 6M guanidine hydrochloride. This seems unfortunate, since the actual portions of crystallographically determined protein structures generally described as coil are not random or disordered in any sense of the word; they are every bit as highly organized and firmly held in place as the repeating secondary structures — they are simply harder to describe. In recent years since recognition of the wide occurrence and importance of tight turns (see Section II,C), they are often separated out as a category of structure, so that now the miscellaneous "coil" category would refer to what is neither helix, β, nor turn.
Turns and bulges are nonrepetitive features which are characterized primarily in terms of backbone conformation and backbone hydrogen bonding. However, much coil structure appears to be very strongly influenced by specific side chain interactions. These have not been very widely analyzed, but a few examples can illustrate the sorts of patterns to be expected. Probably the earliest notice of such a feature is the observation in Kendrew et al. (1961) that serine or threonine frequently hydrogen-bonds to a backbone NH exposed at the beginning of an α-helix. Figure 53 illustrates such a conformation. [This is now called a helix N-cap (Richardson, 1988) and is discussed in section II, A.] Energy calculations for side chain interactions have, quite understandably, considered only the very local region (e.g., Anfinsen and Scheraga, 1975). Recently there have been some systematic empirical computer surveys of side chain environments, such as in Warme and Morgan (1978) and Crippen and Kuntz (1978), and surveys of side chain conformations, such as Janin et al. (1978) and Bhat et al. (1979). These do not focus specifically on nonrepetitive structure, but any strong preferences would be especially influential there. So far, however, these studies are still at the initial stages of tabulating raw statistical preferences. [Later work has tabulated the preferred conformations of side-chain "rotamers" (e.g., Lovell et al., 2000), including the preferred patterns of local sidechain-backbone H-bonds.] (ref ??)
Asparagine position in β sheets | |||
At end of strand |
On a side strand |
In middle of sheet | |
Antiparallel |
30 |
17 |
1 |
Parallel |
10 |
4 |
3 |
a For all residues in β sheet, approximately 50% are in the middle and 50% at either an end or a side. In contrast, less than 20% of the asparagines in parallel sheet are in the middle, and only 2% of those in antiparallel sheet are in the middle. | |||
At the other extreme, it is possible to examine individual examples of a single potentially interesting type to see simply what features turn up. Asparagine is one such potentially interesting residue, since it combines a side chain that mimics a backbone peptide, along with the conformational constraints of possessing only two side chain variable angles. Asn is more likely than any other non-glycine residue to have φ,ψ angles outside the normally allowed regions. Also, it does indeed show a pattern of hydrogen-bonding that is distinct from either glutamine or aspartate. Asparagine is more than twice as likely as any other residue to bond to the first exposed backbone NH or CO at the end of an antiparallel β strand (as in, for example, Fig. 53) and probably for that reason is essentially forbidden from occurring in the interior of antiparallel β sheet although it is very common at or just beyond the edges (see Table I). Asparagine also shows a favored type of hydrogen bond from its side chain CO to the backbone NH of residue n + 2. This pattern happens to predispose a type I tight turn at residues n + 1 and n + 2 (see Fig. 54). [This Asn H-bond also itself mimics a tight turn and has been called a pseudo-turn (Ress)] (ref ??); such cases are treated in detail by ?? ref.
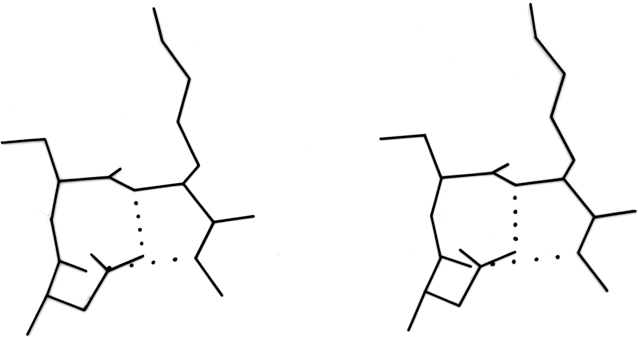
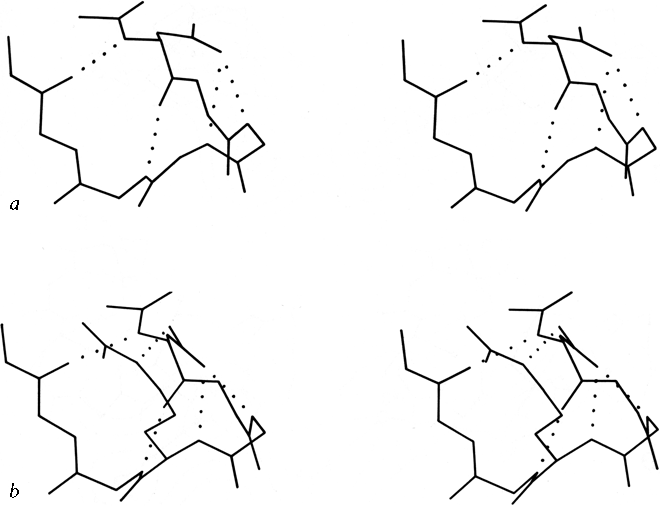
Figure 55a shows a small piece of nonrepetitive structure from pancreatic trypsin inhibitor in which the side chain 0δ of Asn-24 is hydrogen-bonded to both the n + 2 and n + 3 backbone NH groups. There is a G1 β bulge (see Section II,D) at Gly-28, Leu-29, Asn-24. The corner between the two strands is turned by what could be better described as a five-residue turn with one α-helical hydrogen bond and an Asn stabilizing the loop NH groups, rather than as two successive non-hydrogen-bonded turns (which is how it shows up in any computer search for tight turns). An extremely similar conformation occurs in prealbumin (Blake et al., 1978), with the same G1 bulge and five-residue "α" turn stabilized this time by Asp-18 instead of an asparagine, and with an additional bond to the Nε of Arg-21 (see Fig. 55b).
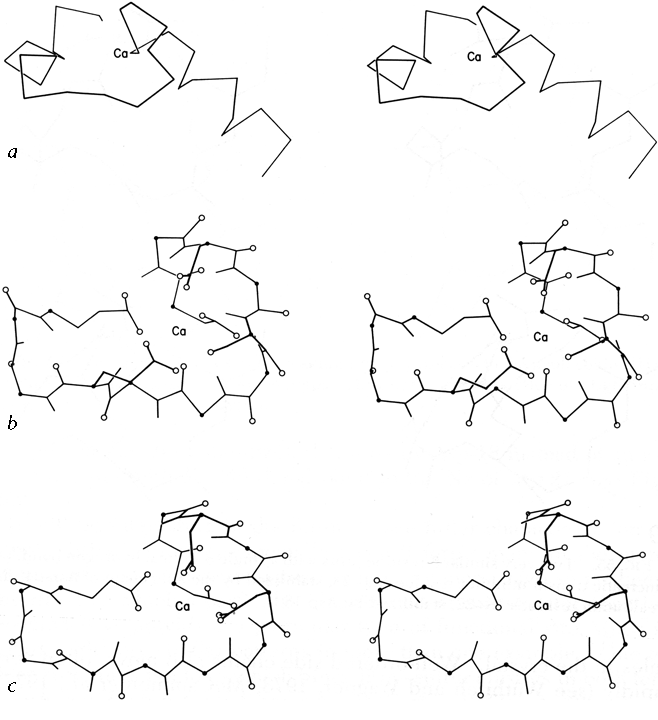
It will be interesting in the future to see fuller characterization of nonrepetitive structure, especially since it forms many of the more complicated enzyme active sites. A few instances have so far been described in which a particular organization of coil structure can be recognized as providing a particular type of functional site, since it occurs with very similar patterns in more than one example. One of these structures is a loop which binds iron-sulfur clusters in both ferredoxin and high-potential iron protein (Carter, 1977). Another such structure is the central loop portion of the "E-F hands" that form the calcium-binding sites in carp calcium-binding protein (Kretsinger, 1976). The backbone structures curl around in very similar conformations and provide ligands in a definite order to the six octahedral coordination sites around the Ca(2+), as illustrated in Fig. 56.
In contrast to the well-ordered but nonrepetitive coil structures, there are also genuinely disordered regions in proteins, which are either entirely absent on electron density maps or which appear with a much lower and more spread out density than the rest of the protein. The disorder could either be caused by actual motion, on a time scale of anything shorter than about a day, or it could be caused by having multiple alternative conformations taken up by the different molecules in the crystal. Well-ordered side chains also move, often very rapidly (see Wüthrich and Wagner, 1978; McCammon et al., 1977), but the movements are brief departures from a single stable conformation.
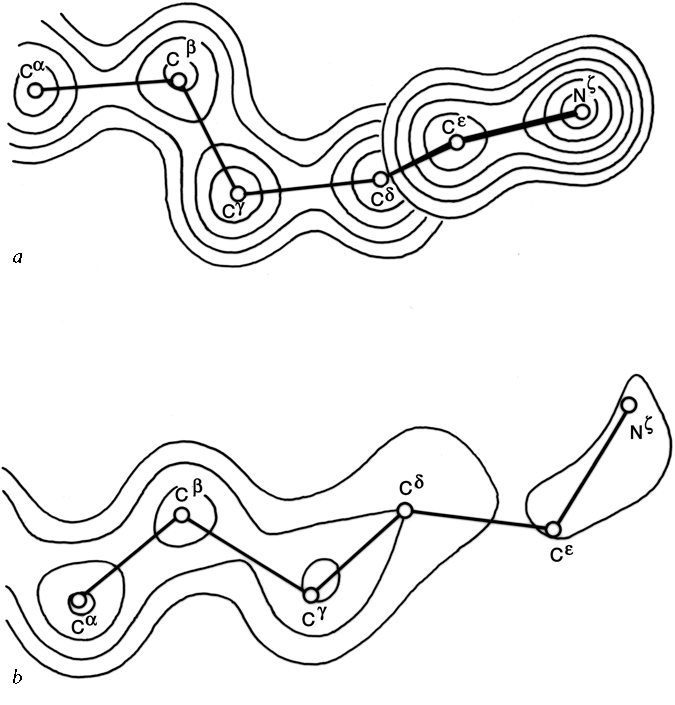
One simple case of disordered structure involves many of the long charged side chains exposed to solvent, particularly lysines. For example, 16 of the 19 lysines in myoglobin are listed as uncertain past Cδ and 5 of them for all atoms past Cβ (Watson, 1969); for ribonuclease S Wyckoff et al. (1970) report 6 of the 10 lysine side chains in zero electron density; in trypsin the ends of 9 of the 13 lysines refined to the maximum allowed temperature factor of 40 (R. Stroud and J. Chambers, personal communication); and in rubredoxin refined at 1.2 Å resolution the average temperature factor for the last 4 atoms in the side chain is 9.2 for one of the four lysines versus 43.6, 74.4, and 79.3 for the others. Figure 57 shows the refined electron density for the well-ordered lysine and for the best of the disordered ones in rubredoxin. Interestingly, arginine side chains do not follow this same pattern. In the structures quoted above, 70% of the arginines were well ordered, as opposed to only 26% of the lysines. In refinement at very high resolution it is sometimes possible to express a partially disordered side chain as a mixture of two different specific conformations, as, for instance, isoleucines 7 and 25 in crambin (Hendrickson and Teeter, 1981). [Alternate conformations have become much more routinely identified now that more structures are solved at very high resolutions. In crambin at 0.54 Å resolution (1EJG), many sidechains, and even parts of the backbone, have alternate conformations, while some sidechains are clearly seen in three different conformations.]
It is fairly common for a few residues at the N-terminus or the C-terminus of the polypeptide chain to be disordered, if they include no large hydrophobic residues. In some cases it is known that these dangling ends can be cleaved off without any loss of stability or activity in the protein (e.g., Anfinsen et al., 1971). However, there are certainly other cases in which disordered regions have very definite functional roles. Sometimes there is disorder in one state and an ordered conformation in another state, with the contrast between the two having a functional role; for example, the inter-subunit salt linkages in hemoglobin which provide constraints in the deoxy form and are free and disordered in the oxy- form (Perutz, 1970, 1978). In systems designed for specific proteolytic cleavage, disorder is one way of promoting cleavage at a given loop. The new chain ends liberated by such cleavage are also very often disordered, as for instance in chymotrypsin (Birktoft and Blow, 1972). In some cases a ligand-binding site may show partial or complete disorder in the absence of the ligand but become well-ordered when the ligand is bound, as for instance the RNA site of tobacco mosaic virus protein (Butler and Klug, 1978). [In triose phosphate isomerase a partially disordered loop over the active site region has, surprisingly, a highly conserved sequence. It turns out this loop becomes ordered on substrate binding, protecting the reaction from solvent and cleverly trading off a loss of entropy against a gain in binding enthalpy to give high specificity without a very tight binding constant that would hinder product release (Petsko).]
One of the most intriguing recent examples of disordered structure is in tomato bushy stunt virus (Harrison et al., 1978), where at least 33 N-terminal residues from subunit types A and B, and probably an additional 50 or 60 N-terminal residues from all three subunit types (as judged from the molecular weight), project into the central cavity of the virus particle and are completely invisible in the electron density map, as is the RNA inside. Neutron scattering (Chauvin et al., 1978) shows an inner shell of protein separated from the main coat by a 30-Å shell containing mainly RNA. The most likely presumption is that the N-terminal arms interact with the RNA, probably in a quite definite local conformation, but that they are flexibly hinged and can take up many different orientations relative to the 180 subunits forming the outer shell of the virus particle. The disorder of the arms is a necessary condition for their specific interaction with the RNA, which cannot pack with the icosahedral symmetry of the protein coat subunits.
Although disordered structure is fairly common in the known protein structures, this is undoubtedly one of the cases in which the process of crystallization induces a bias on the results observed. Since extensive disorder makes crystals much harder to obtain, it seems probable that disordered regions are even more prevalent on the proteins that do not crystallize.
In a very real sense, the structure of the closely bound water molecules around a protein are a part of the protein structure: they determine conformation of the exposed side chains, stabilize the ends of secondary structures, and occupy positions at active sites where they influence substrate binding and sometimes catalysis. The properties of the bulk water are critical in stabilizing the folded native form of proteins (e.g., Kuntz and Kaufmann, 1974), but it is only the bound water that we will consider to be an actual part of, rather than an influence on, the protein structure.

In high-resolution X-ray structures of proteins it is usual for a small number of solvent molecules to appear fairly clearly as peaks in the electron density map (see Fig. 13). Now that various refinement techniques are being applied to many protein structures, determination of water positions is usually a part of the process. In only a few cases, such as the study of rubredoxin in Watenpaugh et al. (1978) and the study of actinidin in Baker (1980), has a real attempt been made to locate all of the fairly tightly bound waters and to eliminate spurious peaks. Figure 58 shows the waters around rubredoxin. Occupancies as well as positions are refined so that partially ordered as well as tightly bound water can be located. It is in fact only relatively few waters for which the occupancy approaches 1 (23 of the 130 waters located in rubredoxin had occupancies ≥ 0.9). [Identification and refinement of waters is now quite standard, and except at low resolution they would usually be assigned in numbers on the order of one per amino acid residue. Note, however, that occupancies and B-factors cannot really be cleanly separated for waters; in some structures these days only B is refined and all waters are left at 1.0 occupancy, but this does not of course mean that they are always present.]
Another recent study which provides less direct, but also very detailed, information about the water around a protein is the Monte Carlo calculations performed by Hagler and Moult (1978) for egg lysozyme. From random starting positions they obtain a very long series of possible sets of water positions for which the statistical properties must obey all the constraints of the energy functions used. Contour maps can be plotted giving the overall frequency of water location at each point, and they match the refined X-ray electron density contours quite well. Also, individual sets of positions at single cycles in the simulation can be examined. The energies of water molecules in various types of locations can be determined for the overall simulation and can be compared with the energy distribution for the bulk water.
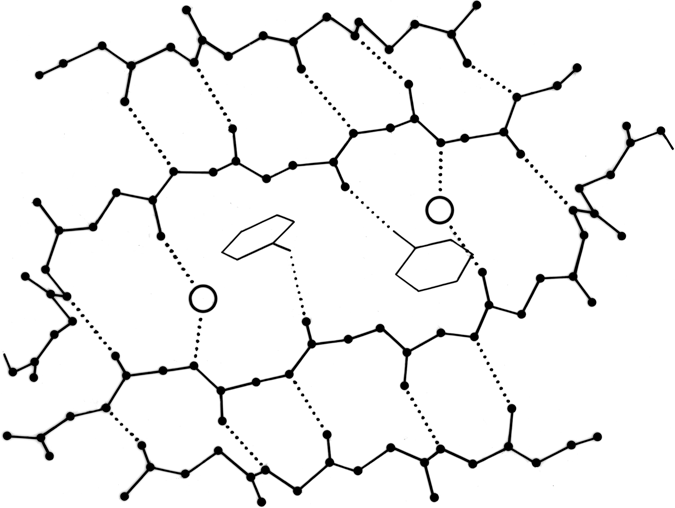
The detailed study of water structure around proteins is only just beginning, but a number of conclusions can be drawn from the crystallographic and theoretical work that has already been done. Isolated water molecules occur trapped inside protein interiors, where they can fill defects in the side chain packing and usually make some hydrogen bonds to protein atoms. Their energies are rather high, but it is much better to have a water than an empty hole in those locations. The number of such internal waters varies very widely from one protein to another. Both for internal and for surface waters, it is very common that they bond to the first free backbone NH or CO groups at the ends of pieces of secondary structure; for β strands it is common that the last H-bond opens up wider, with a water bridging in between (see Fig. 59).

The most ordered surface waters are those around charged side chains or in surface crevices. Occasionally those crevices can be very deep, such as the active site pocket in carbonic anhydrase, which extends about 15 Å in from the surface, with a network of water molecules (Lindskog et al., 1971). The well-ordered waters at the protein surface are usually part of an approximately tetrahedral (but sometimes planar trigonal) network of hydrogen bonds to the protein and to other waters. An example from rubredoxin is shown in Fig. 60.
Both crystallographically and also from vapor-pressure measurements of solvent stabilization (Wolfenden, 1978) it appears that water hydrogen bonds more frequently and more strongly to peptide CO groups than NH groups. In rubredoxin, only 24% of the available backbone NH groups are bonded to water and 70% to other protein atoms, while for the CO groups 43% bond to waters, 41% to protein atoms, and another 8% to both (Watenpaugh et al., 1978).
Many of the tightly bound waters have energies substantially lower than the bulk water (Hagler and Moult, 1978). All studies have found that most of the bound waters, and all of the highly ordered ones, are in the first coordination layer, but that they do not by any means cover the whole protein surface. A substantial number of partially ordered waters are found in the second coordination layer (where they hydrogen-bond to the protein only through first-layer water) and essentially none any further out than that, even where there are suitably sized channels between neighboring protein molecules. The degree of motion seen for individual water molecules also increases dramatically as a function of their distance out from the protein. [Waters, and exposed side chains as well, are more ordered at the cryogenic temperatures now standardly used for data collection, and they are also more ordered in crystal contacts. The molecular contacts in a crystal are usually rather weak, with relatively sparse contact between protein atoms but many contacts through single ordered waters. Such contacts can even have hydrophobic atoms on both sides with rings or clusters of waters in between, as seen in the very high-resolution crambin structure (Teeter).]
Many protein molecules are composed of more than one subunit, where each subunit is a separate polypeptide chain and can form a stable folded structure by itself. The amino acid sequences can either be identical for each subunit (as in tobacco mosaic virus protein), or similar (as in the α and β chains of hemoglobin), or completely different (as in aspartate transcarbamylase). The assembly of many identical subunits provides a very efficient way of constructing large structures such as virus coats. Often a multisubunit molecule is more smoothly globular than its component subunits are, as for instance in the insulin hexamer shown in Fig. 61.
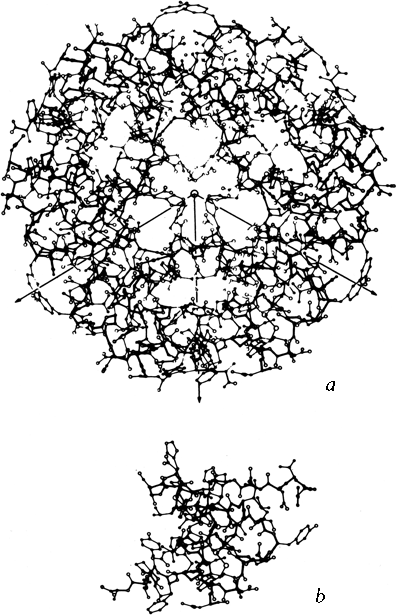
The surfaces that form subunit-subunit contacts are very much like parts of a protein interior: detailed fit of generally hydrophobic side chains, occasional charge pairing, and both side chain and backbone hydrogen bonds. Twofold symmetry is the most common relationship between subunits. The 2-fold is often exact and can be part of the actual crystallographic symmetry, as for the prealbumin dimer in Fig. 62. However, in many cases (e.g., Tulinsky et al., 1973; Blundell et al., 1972) individual side chains very close to the approximate 2-fold axis must take up nonequivalent positions in order to avoid overlapping (see Fig. 63). Conformational nonequivalence can extend further away from the axis and produce such effects as different binding constants for ligands (e.g., Hill et al., 1972). Tetrahedral 222 symmetry is also common, either with only one or with all three 2-folds exact (e.g., Adams et al., 1969).
A rather common feature of subunit contacts is β sheet hydrogen bonding between strands in opposite subunits. Theoretically the relationship could be a pure translation or a 2-fold screw axis with a one-residue translation (for a pair of parallel strands), but all the known cases of inter-subunit β sheet bonding turn out to be between equivalent strands related by a local 2-fold axis. For hydrogen-bond formation, the 2-fold must be perpendicular to the β sheet, requiring the two equivalent strands to be antiparallel. Those may be the only two β strands (as in insulin, Fig. 63), or they may be part of antiparallel β sheets (as in prealbumin, Fig. 62), or the rest of the sheets may be parallel (as in alcohol dehydrogenase domain 1).
Similar subunit structures can assemble in quite different ways. The Greek key β barrel of Cu,Zn superoxide dismutase assembles back-to-back across a tight, hydrophobic side chain contact, while the Greek key β barrel of prealbumin joins by continuing the β sheet bonding side-by-side. Even for proteins known to be closely related the subunits may associate in nonhomologous ways, such as the monomers versus tetramers in various hemoglobins, or the atypical contact between chains in the Bence-Jones protein Rhe (Wang et al. 1979). The homologous domains in the immunoglobulin chains associate in three quite different types of pairwise contact: the usual "back-to-back" barrel contact of VL and VH (shown in Fig. 101); CL and CH1 barrels contact "front-to-front"; and pairs of CH2 domains are quite widely separated by carbohydrate (Silverton et al., 1977). Of course, the great majority of homologous proteins have homologous subunit contacts, but it seems that even a quite drastic change in subunit contacts is easier to accommodate than major internal rearrangement.
Twofold contacts are self-homologous—formed by equivalent surfaces from each of the participating subunits, while the occasional 3-fold (e.g., bacteriochlorophyll protein), 4-fold (hemerythrin), or 17-fold (tobacco mosaic virus) contact is heterologous—formed by joining two different surfaces. An especially interesting type of heterologous contact has been found in hexokinase (Steitz et al., 1976). It was previously assumed that self-associating arrays with a definite number of subunits had to be related by a closed, point-group symmetry operation in order to avoid producing infinite aggregation (e.g., Klotz et al., 1970). Hexokinase, however, has a 156° rotation plus a 13.8Å translation between subunits, which demonstrated clearly that screw axes are also permissible as long as addition of a third subunit by the same screw operation is blocked by overlap with the first subunit. Such a screw-axis relationship can easily produce very marked nonequivalence between chemically identical subunits. [Now that structures are routinely done for large complexes, rotational symmetry is seen quite often, Three-folds (e.g. membrane porins: 2OMF), four-folds (e.g. K+chain: 1BL8), and five-folds (e.g. choleratoxin: 3CHB) are common, and many others occur such as the seven-folds of Gro-EL (1DER), the nine-folds of the light-harvesting complex (1LH2), and the II-folds of TRAP protein (1C9S). In addition to the icosahedral viruses, many other complexes have cubic symmetries, such as ferritin (1AEW) or glutamine synthetase (1FS2).]
Subunit contacts need to be relatively extensive and stable if they are to ensure subunit association in the absence of a covalent link. However, in some cases a subunit contact can shift back and forth between two different stable positions, as has been demonstrated for oxy- versus deoxyhemoglobin (Perutz, 1970). Allosteric control can then be exerted by any factors which either affect the local conformation or bind between the subunits. A less elegant but even more extreme example is lamprey hemoglobin, which dissociates altogether in the oxy form (Hendrickson and Love, 1971).
Subunit motion between two positions is also critical to the assembly of tobacco mosaic virus. In the partially assembled "disks," having two stacked layers of 17 subunits each, the layers are wedged apart toward their inner radius. During assembly of the viral helix, RNA binds between the layers, which then clamp tightly together with 16(1/3) subunits per turn (Bloomer et al., 1978; Butler and Klug, 1978).
Within a single subunit, contiguous portions of the polypeptide chain frequently fold into compact, local, semi-independent units called domains. The separateness of two domains within a subunit varies all the way from independent globular domains joined only by a flexible length of polypeptide chain to domains with extremely tight and extensive contact and a smoothly spherical outside surface for the entire subunit (such as in Fig. 64). An intermediate level of domain separateness is common in the known structures, with an elongated overall subunit shape and a definite neck or cleft between the domains, such as phosphoglycerate kinase shown in Fig. 65.
Another feature frequently seen in both domain and subunit contacts is an "arm" at one end of the chain which crosses over to "embrace" the opposite domain or subunit. Figure 66 shows such "arms" on the domains of papain. "Arms" that cross between domains or subunits almost invariably lie at the surface, but one unusual case in influenza virus hemagglutinin has a piece from a different domain forming the central strand of a five-stranded β sheet (see Fig. 83, where the alien strand is shown by the dotted lines).

The paucity of examples of flexibly hinged domains is almost certainly due to the difficulties of crystallizing such structures. In the immunoglobulins it has long been known from electron microscopic and hydrodynamic evidence that the hinge between the Fab and Fc regions is very flexible. The intact Dob immunoglobulin whose structure has been determined (Silverton et al., 1977) has a substantial deletion in the hinge region which presumably limits its flexibility greatly. Intact immunoglobulins without such a deletion are notoriously difficult to crystallize, and the two cases in which crystallization has succeeded both turned out to have ordered Fab regions and invisible, disordered Fc portions (Colman et al., 1976; Edmundson, 1980). A study of diffracted X-ray intensity as a function of resolution has shown that the Fc disorder is probably a static, statistical disorder among at least four multiple conformations (Marquart et al., 1980). [More recently, two more intact immunoglobulin structures have been solved, one hinged into a Y shape (1IGY) and the other a T shape (1IGT). ] This motion of flexibly-linked domains is illustrated in Fig. 66xhe associated kinemage ?? BnT c15,k2.

At the other extreme, with very tightly associated domains, it is rather difficult to make the decision as to how many domains should be said to be present. In naming domains for the present study we have made use wherever possible of experimental evidence about either hinge motions of domains or about their folding or stability as isolated units. For example, rigid-body hinging has been documented for hexokinase (Bennett and Steitz, 1978), phosphoglycerate kinase (Pickover et al., 1979), tomato bushy stunt virus (Harrison et al., 1978), and between immunoglobulin VL and CL domains (Schiffer et al., 1973; Abola et al., 1980). On the other hand, the known movements on substrate binding in carboxypeptidase (Quiocho and Lipscomb, 1971), adenylate kinase (Sachsenheimer and Schulz, 1977), and phosphorylase (Sygusch, et al., 1977) involve only surface movement of loops. For elastase (Ghelis et al., 1978) it is known that after proteolytic cleavage the domains can fold up into stable isolated units. But on the other hand, none of the large fragments of staphylococcal nuclease (Taniuchi and Anfinsen, 1969) or of cytochrome c (Fisher et al., 1973) can fold up independently. For the majority of proteins, where such experimental evidence is not available, the decision about domains was made on the basis of analogy: whether the whole subunit or its parts more closely resembled other single-domain proteins. It is possible that some of the larger domains listed here will turn out to have genuinely independent smaller parts; domain divisions have been claimed within subtilisin (Wright et al., 1969; Honig et al., 1976) and the first domain of phosphorylase (Weber et al., 1978). Several domains of more than 300 residues are now known, however, which cannot plausibly be subdivided [e.g., triosephosphate isomerase, carboxypeptidase, bacteriochlorophyll protein]. Since large domains are clearly possible, we have been conservative in assigning divisions. Only three of the domains consist of two long, noncontiguous segments (e.g., pyruvate kinase d1); most are a single piece of chain.
The above definition of domains, in which they are thought of as potentially independent, stable folding units analogous to subunits, is only one of three rather separate concepts of domains in current thinking about the subject. The initial recognition of domain divisions as a general feature in the three-dimensional structures (Wetlaufer, 1973) was in terms of locally compact globular regions also contiguous in the sequence. This idea has recently been elaborated and computerized in terms of maximum long-range contacts between segments with only short-range internal contacts (Crippen, 1978), binary divisions of the sequence that maximally lie on opposite sides of a single cutting plane (Rose, 1979), or binary sequence divisions that minimize the sum of the separate surface areas (Wodak and Janin, 1980). Each of these algorithms agrees in many cases with "intuitive" or "subjective" division into domains, but one of the more striking results of this kind of analysis is that it always produces a hierarchy or tree of substructures, usually containing two or three levels between the individual secondary-structure elements and the entire subunit. Domains as usually conceived represent the upper levels of such a hierarchy, while the lower levels may very well represent intermediates in the folding process. One lesson from these studies, however, is that something else is involved in the intuitive concept of a domain besides these purely geometrical criteria of relative compactness. In the current study we have provided that additional criterion by requiring analogy to some structure known to possess stability in isolation.
The third major concept of domains is basically genetic, following quite naturally from the earliest idea of domains: the homologous, internally disulfide-linked regions in the immunoglobulin sequence (Edelman and Gall, 1969). Rossmann (Rossmann et al., 1974) has proposed that the similar nucleotide-binding domains in various dehydrogenases represent genetic segments that have been transferred and combined with differing catalytic domains to produce functionally distinct but partially related enzymes. Recently the discovery of exon (translated and expressed) DNA sequences separated by intron sequences which are clipped out of the mRNA before translation into protein has raised the intriguing possibility that separate exons correspond to structural and/or functional domains recognizable in the proteins and basic to their evolutionary history. In immunoglobulins the exons correspond quite exactly with structural domain divisions, with the hinge region as a separate exon (Sakano et al., 1979). However, in hemoglobin the introns occur not only inside what is usually considered a single domain, but even in the middle of individual helices, although it has been found that the isolated central exon peptide can bind heme (Craik et al., 1980). At the current level of knowledge it is unclear whether exons will provide a clarification of the basis of structural domains, although they are clearly a fundamental breakthrough in our understanding of the evolutionary processes involved.
Domains have proved so fruitful in explaining both the structure and the function of proteins that the concept is certain to survive in one form or another. At some time in the near future it will presumably acquire full scientific respectability with a verifiable definition in terms of either folding units or genetic units or perhaps both. [In practice, there are now two accepted standards at opposite ends of the domain problem. Genomics and bioinformatics now allow routine, reliable identification of functional domains that recur in many different sequence contexts, often as repeats but occasionally as isolated subunits; a few examples are zinc fingers, pleckstrin homology domains, and fibronact in type 3 domains. At the other extreme, of difficult and perhaps unique cases in individual structures, the de facto standard is the assignments made by Alexci Murzin for the SCOP database (http://scop.mrc-lmb.ac.uk/scop).]

The domains (as we have defined them) within a subunit very often resemble each other (as discussed in Section IV,B), and those similar domains are frequently related by an approximate 2-fold axis. The 2-fold relationship often occurs even in cases in which it involves considerable inconvenience because the start and end of the chain are on opposite sides of the basic domain structure, so that a long additional loop is required to connect the end of the first domain to the beginning of the second one (see for instance Fig. 67). An even more interesting instance of the pervasiveness of 2-fold domain contacts is in the serine proteases, where the relationship between genetically equivalent portions of the chain is in fact a pure translation. However, the initially heterologous contact seems to have evolved toward 2-fold similarity of the contact surfaces, so that now these proteins give the appearance of having a 2-fold relationship around the midpoint of the sequence (see Fig. 68). This convergent evolutionary process was able to produce an apparent sequence inversion because the topology of the serine protease domains (+1, +1, +3, -1, -1, or -1, -1, -3, +1, +1) is invariant to reversal of N- to C-terminal direction. The kinds of cases illustrated in Figs. 67 and 68 suggest that 2-fold contacts are inherently easier to design well than unmatched, heterologous contacts.
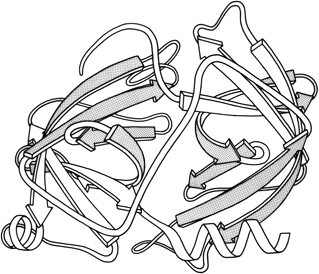
The types of contacts between domains are in general very similar to those found between subunits, but there are a few characteristic differences. Continuation of β sheet hydrogen-bonding, which is very common between subunits, is almost unknown between domains (which typically contact through side chains rather than main chain). β barrel domains (see Section III,D) most often associate side-by-side, usually with their barrel axes at a 50 to 90° angle to each other (as in Fig. 68). Doubly wound parallel β sheet domains (see Section III,C) most often associate with the β strands pointing toward each other (as in Fig. 68).
Domains as well as subunits can serve either as moving parts for the functioning protein or as modular bricks to aid in efficient assembly. Undoubtedly the existence of separate domains is important in simplifying the folding process into separable, smaller steps, especially for very large proteins. The commonest domain size is between 100 and 200 residues, but it now appears that there is no strict and definite upper limit on practical folding size: domain sizes vary by an order of magnitude, from 41 residues up to more than 400. The range of domain sizes is somewhat different for each of the major structural categories (see Sections III,B through III,E for definitions): generally less than 100 residues for small disulfide-rich or metal-rich proteins, 80 to 150 for antiparallel α, 100 to 200 for antiparallel β, and 120 to 400 for parallel α/β. The lower limit for each category presumably reflects the smallest stable structure that can be made using that general design pattern. The upper limit may, among other things, reflect the largest domain for each structure type that can fold up efficiently as a single unit.
The other important function of domains is to provide motion. Completely flexible hinging would be impossible between subunits because they would simply fall apart, but it can be done between covalently linked domains. More limited flexibility between domains is often crucial to substrate binding, allosteric control, and assembly of large structures. In hexokinase the two domains hinge together on binding of glucose, enclosing it almost completely (Bennett and Steitz, 1978) [see kinemage ??.] . Not only does this mechanism provide access to a very tight and hydrophobic specificity site, it has also been hypothesized as necessary in order to discriminate against using water as a substrate and acting as a counterproductive ATPase. It should be remembered, however, that domain motion is not the only way to solve the problem: what hexokinase and alcohol dehydrogenase accomplish by domain hinging, adenylate kinase and lactate dehydrogenase accomplish by large movements of surface loops. [Domain hinge motion has remained extremely common, especially for enzymes, many of which show the overall pattern of two lobes with the active site in a cleft between them.]
In tomato bushy stunt virus protein, domain hinging helps to solve the problem of packing 180 identical protein subunits quasi-equivalently into the 60-fold icosahedral symmetry of the virus shell (Harrison et al., 1978). Around the exact 2-fold and the quasi-2-fold axes of the icosahedron, contact must be made between chemically identical subunits which come together at rather different angles around the two kinds of 2-fold axes. Identical contacts are made in both cases between pairs of protruding d3 domains, but the angle between d2 and d3 domains can change by about 20° (see Fig. 69) so that each type of d2 domain is placed in the correct orientation to interact around the 5-fold or the quasi-6-fold axes. Pairs of d2 shell domains form two different types of contacts: in one type the two surfaces are tightly packed and the N-terminal arms are disordered, while in the other type of contact the N-terminal arms of "C" subunits fill a wedge-shaped opening between the contact surfaces and then wind around the 3-fold axis (see Figs. 70 and 84).
Having looked at the characteristics of individual structural features and some of their local combinations, we are now in a position to sort out and classify the major structural patterns, or "folds," that make up entire proteins. This classification will build on earlier work by Rossmann (e.g., Rossmann and Argos, 1976), Richardson (1977), and Levitt and Chothia (1976), but will attempt to combine and extend those systems, as well as including the newer structures now available.
[In 2006, the major resources for classification of protein structures are SCOP (scop.mrc-lmb.cam.ac.uk/scop/) (Murzin, 1995) and CATH (www.cathdb.info/latest/index.html) (Orengo, 1997) which use major categories similar to the ones here: a lowest “family” level from sequence homology, and distinct versions of “fold”-based categories in between. Both are organized by domains. Further notes on the many new types of folds seen since 1981 will be found in the later sections.]
The most useful level at which to categorize protein structures is the domain, since there are many cases of multiple-domain proteins in which each separate domain resembles other entire smaller proteins. We have separated proteins into domains on the basis of whether the pieces could be expected to be stable as independent units or are analogous to other complete structures (see Section II,I for a fuller explanation). Clearly demonstrated homologous families such as trypsin, chymotrypsin, elastase, and the S. griseus proteases, or the cytochromes c, c2, c550, c551, and C555, are treated as single examples. There are between 90 and 105 different domains represented in the current sampling of known protein structures, depending on how one counts the cases of similar domain structures within a given protein. In the schematic drawings of Figs. 72-86 such domains are illustrated separately only if they are at least as different as the range of variation common within the close homology families (and, of course, if suitable coordinates or stereos were available). The domains within each protein are distinguished by numbers in sequence order (e.g., papain dl and papain d2), except for the immunoglobulins for which we use the standard terminology (VL, CH1, etc.) for constant and variable domains.
Structural categories are assigned primarily on the basis of the type and organization of secondary-structure elements, the topology of their connections, and the number of major layers of backbone structure that are present. Since proteins fold to form a protected hydrophobic core of side chains on the interior, the simplest type of stable protein structure consists of polypeptide backbone wrapped more or less uniformly around the outside of a single hydrophobic core. We will describe such a structure as "two layer," because a line from the solvent through the center of the protein and back out again would pass through two principal layers of backbone structure (see Fig. 71a). Over half of the known domain structures have two layers. About a third of the structures have three layers of backbone and two hydrophobic cores; the commonest such type has a central β sheet layer flanked by two helical layers (see Fig. 71b). There are three known four-layer domains (e.g., Fig. 71c) and one five-layer. Isolated loops that curl over the outside are not considered to form a distinct layer. Although there are some ambiguous cases (especially in the very small structures) they are less common than one might expect, presumably because the requirements for rapid folding rule out much tangling or recrossing of the backbone.

The approximately 100 distinctly different domains fall into four broad categories, each of which has several subgroupings. The four broad categories are (I) antiparallel α; (II) parallel α/β; (III) antiparallel β; (IV) small SS-rich or metal-rich. The major determinant in assigning a domain to one of these categories is not just the percentage of a given secondary structure, but whether that type of secondary structure forms the central core and whether its interactions could be the dominant stabilizing ones. The two β categories are the most populous, with 30 to 35 members each; there are about 20 α-helical domains and a dozen of the small proteins. The overall classification scheme is summarized in Section III,A,2. An alphabetical index of the proteins is given in Section III,A,4, with domain assignments, structure subcategories, and literature references.
Obviously this classification is not the only plausible way to categorize protein structures. Indeed, for some of the individual cases there are other descriptions which would be preferable because they emphasize possible relationships to functionally similar proteins. However, the motivation here has been to achieve the most satisfactory compromise for all the structures: to fit as many examples into as few groupings as possible, while retaining enough detail to provide meaningful descriptions. Also, the prejudice has been in favor of grouping together domains whose entire structure is approximately the same rather than cases in which a relatively small portion of both structures is more exactly similar.
In order to illustrate this taxonomic system and to facilitate contrasts and comparisons among the structures, schematic backbone drawings have been made of most of the known structures. The drawings are grouped together by categories in Section III,A,3. α-Carbon coordinates were displayed in stereo on Richard Feldmann's computer graphics system at NIH; a suitable view was chosen (consistent for each subcategory of structure), and plotter output was obtained at a consistent scale (approximately 20Å per inch on the final drawings as reproduced here). The schematic was drawn on top of the plotter output for accuracy, with continual reference to the stereo for the third dimension. Loops, and to some extent β strands, were smoothed for comprehensibility, and shifts of 1 or 2Å were sometimes necessary in order to avoid ambiguity at crossing points. A uniform set of graphical conventions was adopted (see Section III,A,3 for explanation) in which β strands are shown as arrows, helices as spiral ribbons, and nonrepetitive structure as ropes. Location and extent of β strands and helices are sometimes based on published descriptions and hydrogen-bonding diagrams, but often must be judged from the stereo view itself. Very short β interactions are shown as arrows when they form part of a larger sheet but may be left out if they are isolated. Foreshortening, overlaps, edge appearance, and relative size change are used to provide depth cues.
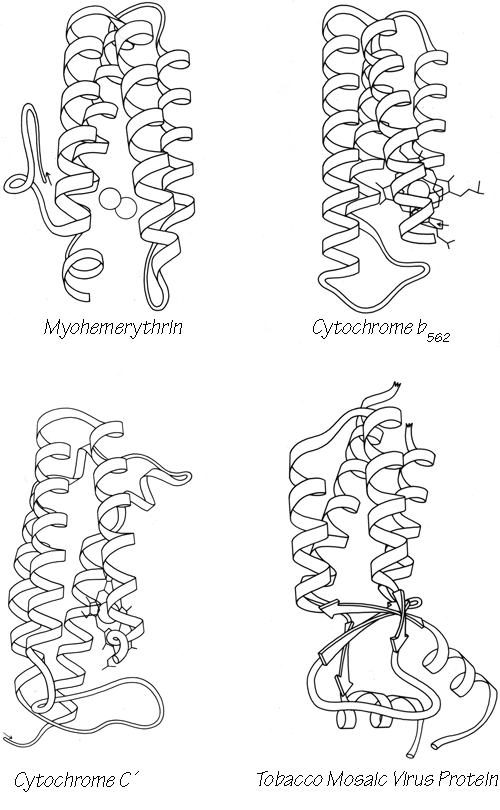
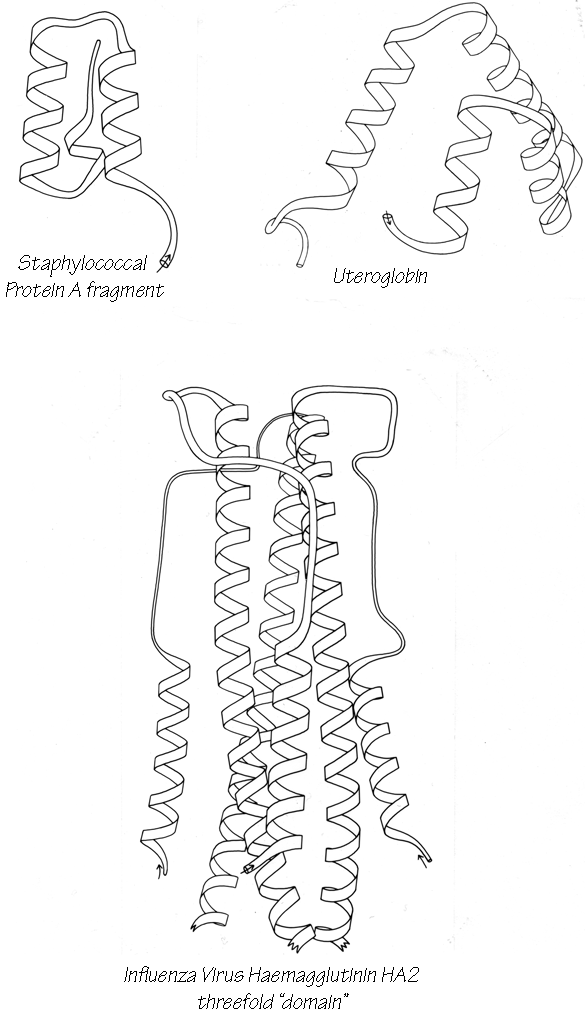
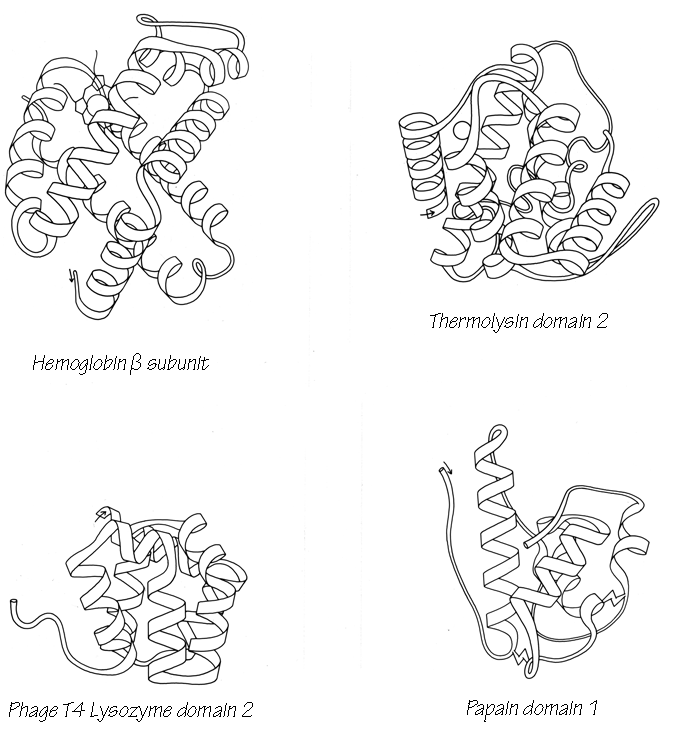
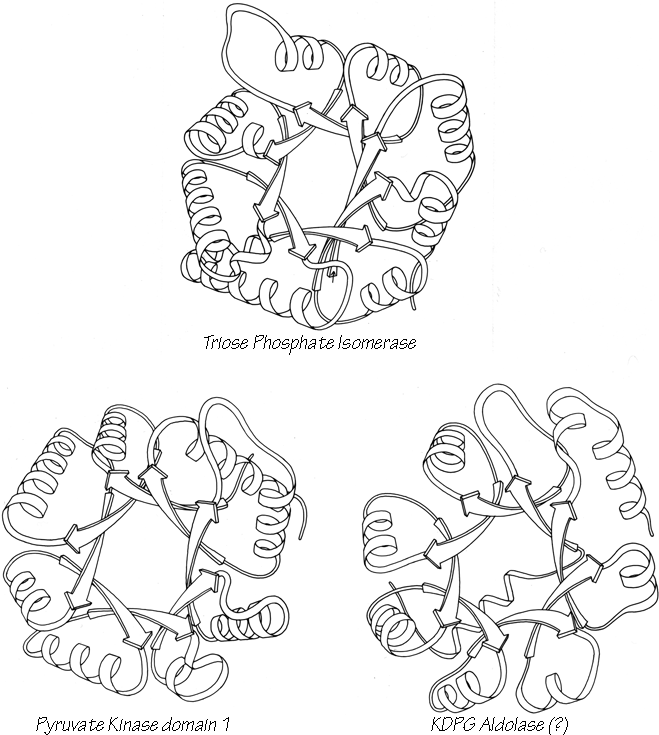
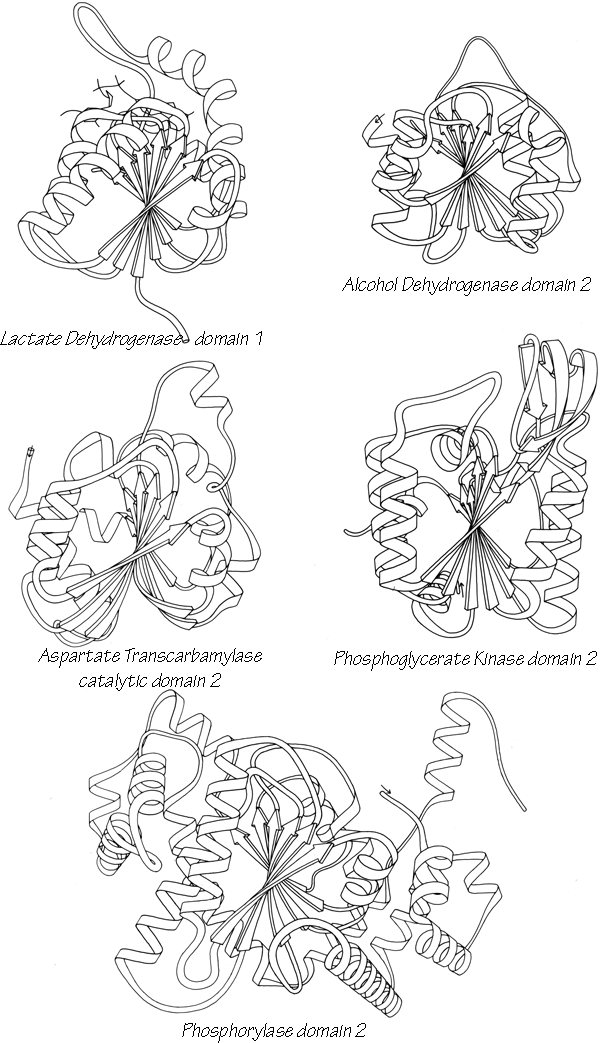
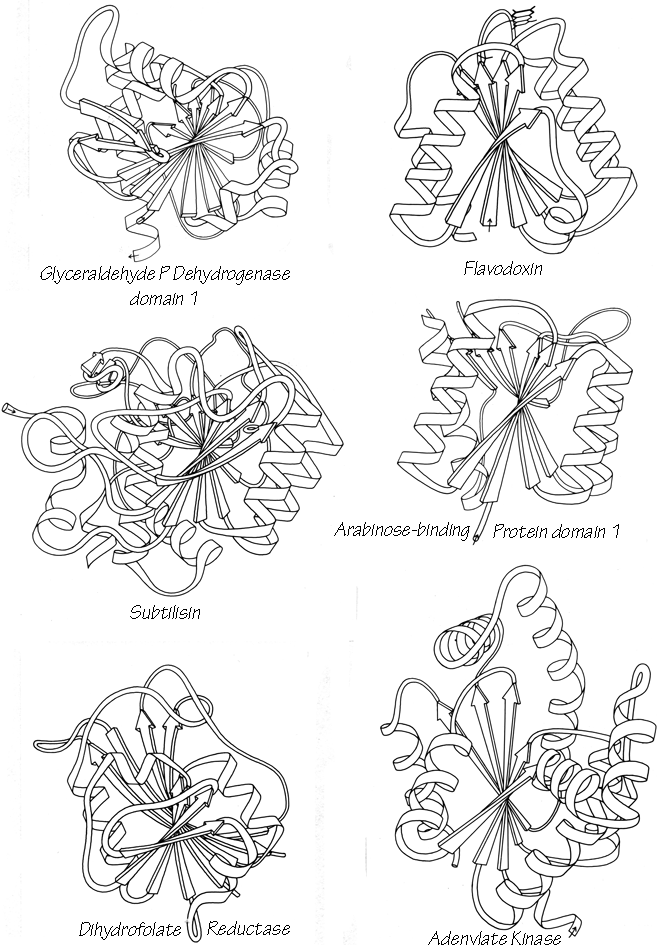
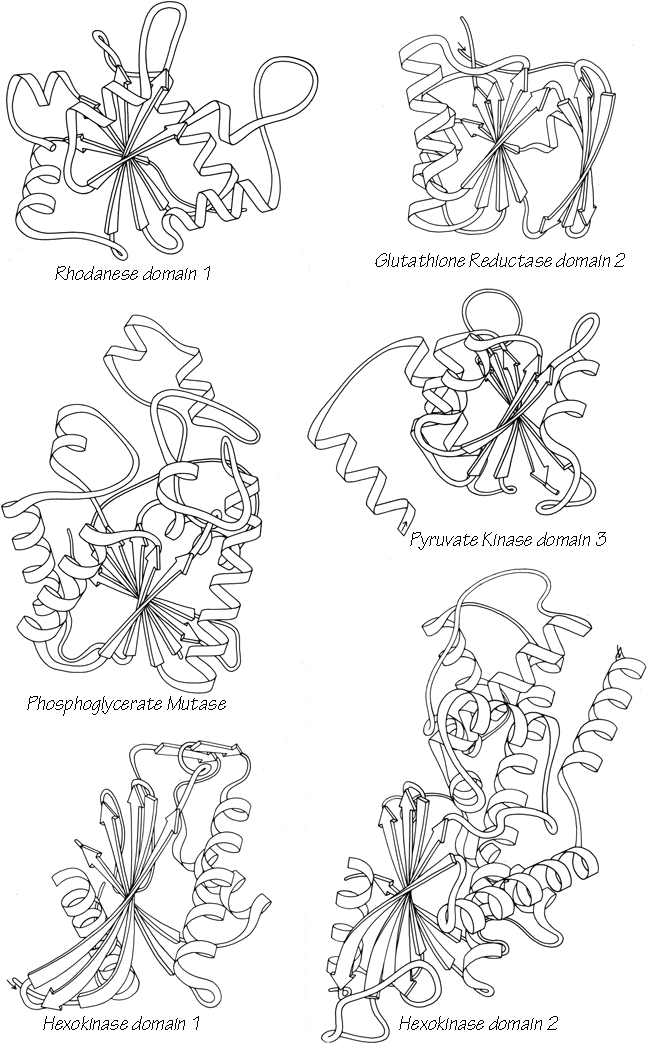

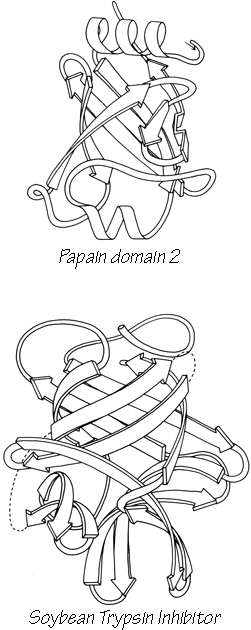
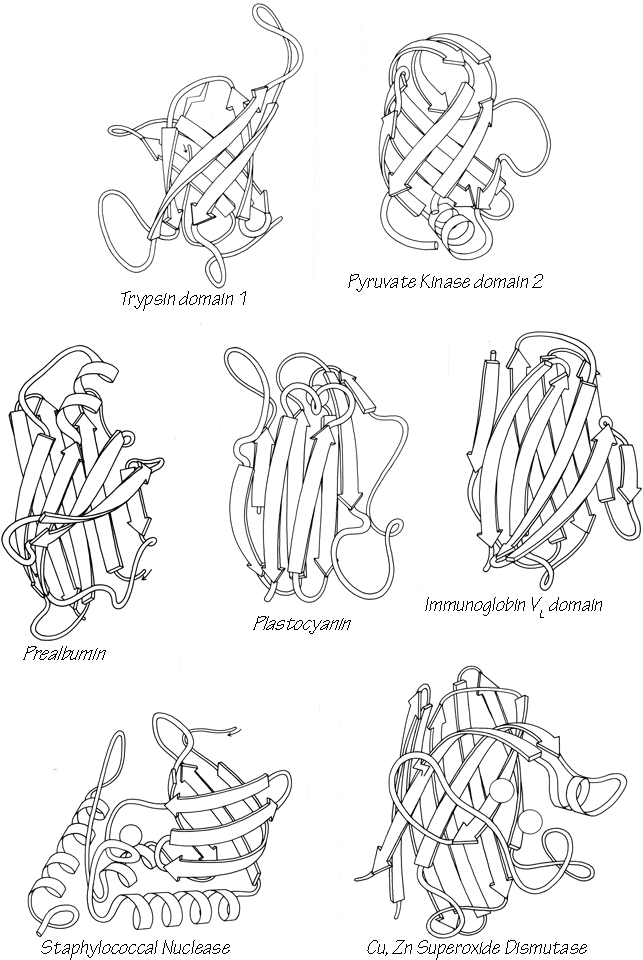
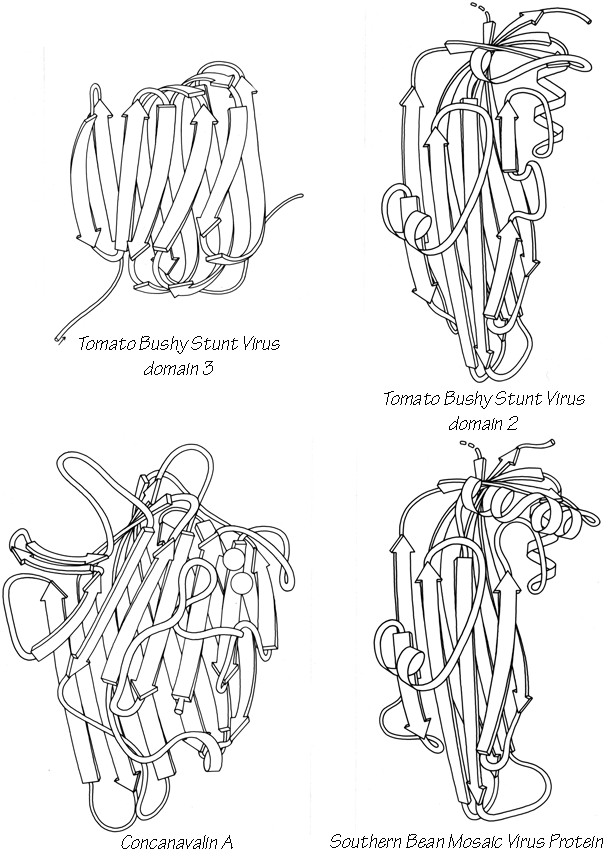
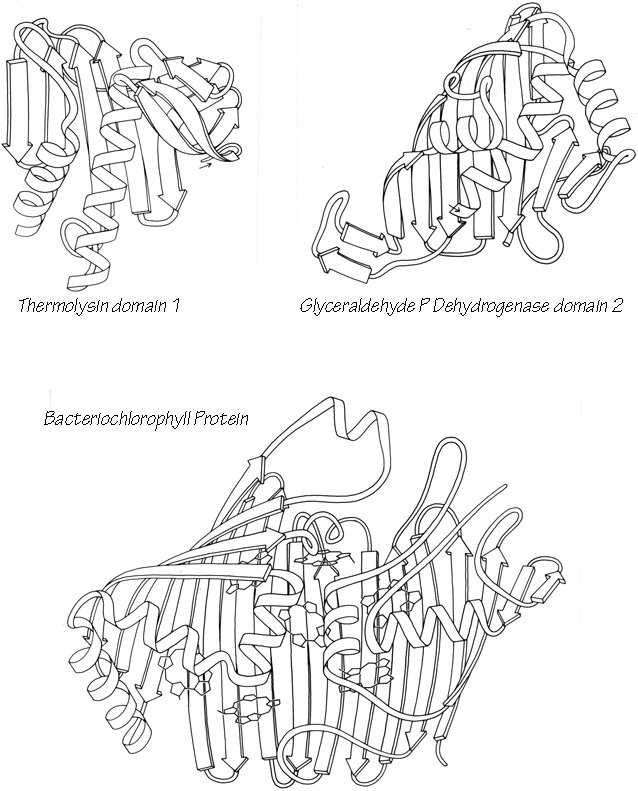

Detailed discussions of the categories and subgroupings are given in Sections III,B through III,E. The scale of these drawings (Figs. 72-86) is approximately 20Å to the inch. β strands are shown as arrows with thickness, helices as spiral ribbons, and nonrepetitive structure as ropes. Disulfides are shown as "lightning bolts." Circles represent metals, and some prosthetic groups are shown as atomic skeletons, but not for all cases in which they are known to be present. A question mark in the label means that backbone connectivity is uncertain in some places. Where needed for clarity, the N-terminus of the domain is indicated by a small arrow; for a few two chain domains the C-terminus is indicated as well. [More detailed information on how the drawings were made is available in a Methods of Enzymology section (J.Richardson, 1985) and in a historical retrospective (J.Richardson, 2000 Nature Str. Biol.), and some related issues about structure representation in a review lecture (Richardson, 1992 Biophys. J.).]
[Protein structure schematics, figures 72 through 86, are presented in the web version as a gallery. That is, each figure is presented individually and navigation buttons to each figure and back to this page are available from each schematic. This allows a larger image for each schematic and faster loading in the browser. Links to each figure are given below:
[PDB (Protein Data Bank) ID codes are given here for all cases where the drawing was based on coordinates then available in the Atlas of Macromolecular Structure on Microfiche (Feldmann, 1977) or in the Protein Data Bank (Bernstein, 1977). For those drawings made from coordinates provided by the crystallographers (see acknowledgements) or for the few made from published stereo figures, the PDB code is given only where that later-deposited coordinate set is essentially the same as the one used.]
The first major grouping of structures contains domains that are essentially all α-helical. Since there is relatively little other structure besides the helices, the simplest ways of connecting them involve predominantly antiparallel helix interactions, and that is in fact what is observed for these proteins. This category corresponds to Levitt and Chothia's all-α category, but it has more members both because of a number of new structures and because of helical domains in proteins they classified as α + β (such as thermolysin). [This catergory is again now called “All-α”, both to match the “All-β” which now has parallel β folds (see below), and also because of the increased role of perpendicular helices (here seen only for the E-F hands of carp Ca-binding protein in Fig. 74) and of multi-chain parallel coils (here seen only for the flu haemagglutinin in Fig. 72).]
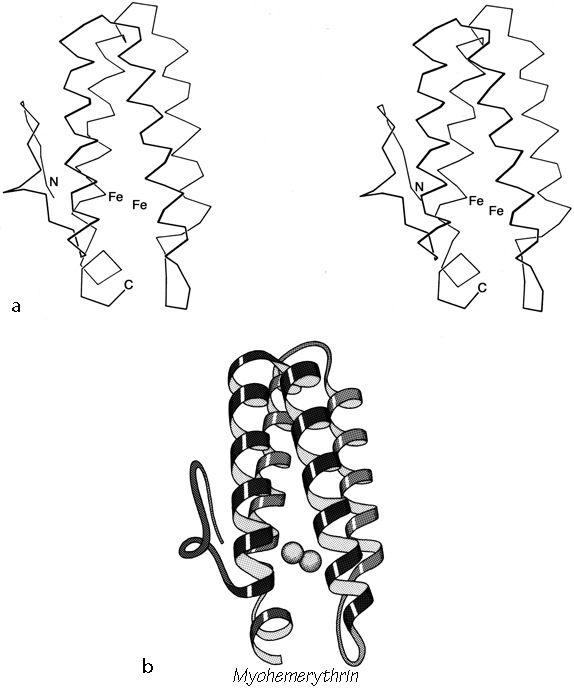
Figures 72 through 74 show schematic diagrams of the antiparallel domains, grouped into subcategories. Almost all of them are two layer structures. The simplest and commonest subgroup looks like a bundle of sticks: usually four helices bundled in a cylinder with simple +1 connections. Most of the helices are quite close to exactly antiparallel, with typically a left-handed superhelical twist of less than 15° relative to the common axis of the bundle. These structures were first described as a group in Argos et al. (1977). Figure 87 illustrates myohemerythrin as an example of this structure type, showing an α-carbon stereo, a schematic drawing, and a topology diagram.
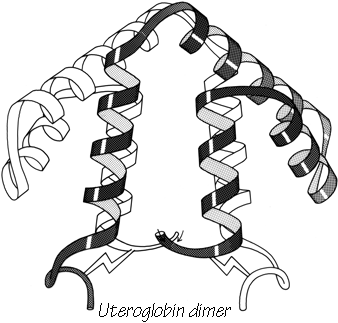
The simple up-and-down helix bundle structures include the hemerythrins (myohemerythrin and the hemerythrin subunits), cytochrome b562, cytochrome c', uteroglobin, tobacco mosaic virus protein, staphylococcal protein A fragment, and probably the ferritin subunits. Tyrosine-tRNA synthetase domain 2 has quite a similar organization, but the last helix tilts away from the bundle (Blow et al., 1977). The uteroglobin subunit also has its fourth helix out to one side, but in the dimer molecule (Fig. 88) those final helices each complete a compact four-helix bundle with the rest of the opposite subunit. In cytochrome c' there is a similar but less extreme arrangement in which the first helix lies at a greater angle to the bundle axis and forms the tightest part of the dimer contact. [The uteroglobin arrangement is an early example of what is now called a domain-swapped dimer (Eisenberg)] (###REF)
Tobacco mosaic virus protein has a small, highly twisted antiparallel β sheet at the base of the helix bundle, with two more helices underneath the sheet (see Fig. 72). Cytochrome b5 looks remarkably similar (see Fig. 105), but the helices are much shorter. That structure could have been classified as an up-and-down helix bundle, but we have placed it in the small metal-rich proteins because its helix bundle is very small and distorted and the heme interactions appear more important than the direct helix contacts.
All but one of the above structures have four helices in the bundle, with + 1,+ 1, + 1 connections. For the up and down topology on a cylinder, handedness can be defined by whether the chain turns to the right or to the left at the end of the first structure element (whether it is a helix or a β strand). With an even number of helices, reversing N to C direction of the chain also reverses handedness of the topology; for an odd number of helices or strands handedness is invariant to chain reversal. For + 1, + 1, + 1 topologies in general, handedness is not a very robust criterion of similarity, since it reverses on addition or deletion of one of the structure elements at the N-terminus but not at the C-terminus, so that a given five-helix structure could have evolved from either handedness of four-helix structure. Hemerythrin, cytochrome b562, cytochrome c', uteroglobin, and tobacco mosaic virus protein are all right-handed, while cytochrome b5, tyrosine-tRNA synthetase, and staphylococcal protein A fragment are left-handed. [The up & down 4-helix bundle is a very common superfold. But more complex topologies with long connections occur] , such as
The connectivity is not known for the seven-helix bundle of purple membrane protein (Henderson and Unwin, 1975), but on the basis of its resemblance to other antiparallel a proteins the most likely topologies would be either up-and-down or Greek key (see below). An analysis based on the sequence and the relative electron-densities of the helices (Engelman et al., 1980) considers a left-handed up-and-down topology as the most probable model. [The up & down 7 helix bundle structure was confirmed by higher resolution electron diffraction and xray structures, both for purple membrane protein (bacteriorhodopsin) and for rhodopsin.]
Many of the up-and-down helix bundle proteins form large multisubunit arrays. Hemerythrin is an octomer, with the end of one helix bundle butting against the side of the next one around the 4-fold axis (Ward et al., 1975). The 24 ferritin subunits form a hollow spherical shell with the helix bundles approximately tangential to the shell and the subunit interactions around the 3-fold and 4-fold axes rather like the interactions in hemerythrin. Tobacco mosaic virus protein, on the other hand, forms a tightly packed long helix of subunits; the a-helical bundles are aligned radially, with RNA bound at their inner ends. Purple membrane protein spans the membrane, forming a two dimensional crystalline array with the helix bundles perpendicular to the membrane and parallel to each other around the 3-fold axis.
One of the most important and interesting antiparallel α structures is the globin fold, which has been found in the three-dimensional structures of a large group of related proteins including myoglobin and the hemoglobins of various mammals, glycera, lamprey, insect and even legume root nodules. The globin fold is a good example of how there may be several alternative useful ways of describing a given structure. To someone studying hemoglobin function the relevant level of description includes all the structural detail that can be made comprehensible, or perhaps generalized to include what is common to all the globin structures. On the other hand, if one is concerned, as we are here, with obtaining a memorably simple description of the whole structure and relating it to other protein structures, then the issue is deciding which features are most important to include in the simplification and with which if any other proteins it can meaningfully be compared. Classifying the globins as all-α proteins is obviously true and useful, but Levitt and Chothia's (1976) scheme of representing the globin topology does not suggest similarities to any of the other all-a proteins, even when the more recent structures are included. Argos and Rossmann (1979) have suggested an interesting similarity of structure around the heme pocket for the globins, cytochrome b5, and cytochrome c551. Their description is probably the most relevant one for trying to understand how heme-binding pockets are organized, but it does not seem suitable as a general structural description since the omitted halves of the three structures are all extremely different and do not form separable domains.
Figure 89 illustrates two different tries at simplified representation of the globin structure. For reference, Fig. 89a shows the hemoglobin β chain in stereo. Figure 89b shows the globin structure schematically as two layers of helices with the elements in one layer approximately perpendicular to those in the other layer; this can be contrasted with a possible description of the up-and-down helix bundles as two layers with their elements approximately parallel to each other. The perpendicular layers provide a rather successful simple schema for the globin structure, but unfortunately there are no other proteins that can be adequately described as two perpendicular layers of helices. Also, specification of the topology in this scheme is cumbersome, since the chain skips back and forth between layers.
Figure 89c schematizes the globin structure as a twisted cylinder of helices, analogous to the antiparallel β barrels to be discussed in Section III,D. The up-and-down helix bundle structures are of course also readily described as cylinders, so that this schema makes the majority of antiparallel α structures directly analogous to the majority of antiparallel β structures. Their topologies can be conveniently specified by the simple nomenclature listing connection types (see Section II,B). The major irregularity of the globin fold when considered as a cylinder is that one element (the A and B helices) bends sharply to close the cylinder; this feature is also seen in five- and six-stranded β barrels such as trypsin. But perhaps the most satisfying feature of schematizing the globin fold on a cylinder is that it can then be grouped with other structures (thermolysin d2, T4 phage lysozyme d2, papain dl, and cytochrome c peroxidase d2) which also show the "Greek key" (see Richardson, 1977) topology of + 3,- 1,- 1. Papain domain 1 also shows the diagnostic feature of Greek key structures by containing a non-nearest-neighbor connection which skips across the end of the cylinder, however, most of its helices are short and they form a rather irregular bundle. Papain domain 1 contains two disulfides; we will find repeatedly that increasing disulfide content goes along with decreasing regularity of both secondary and tertiary structure. [Even more globin-fold-like is ###]
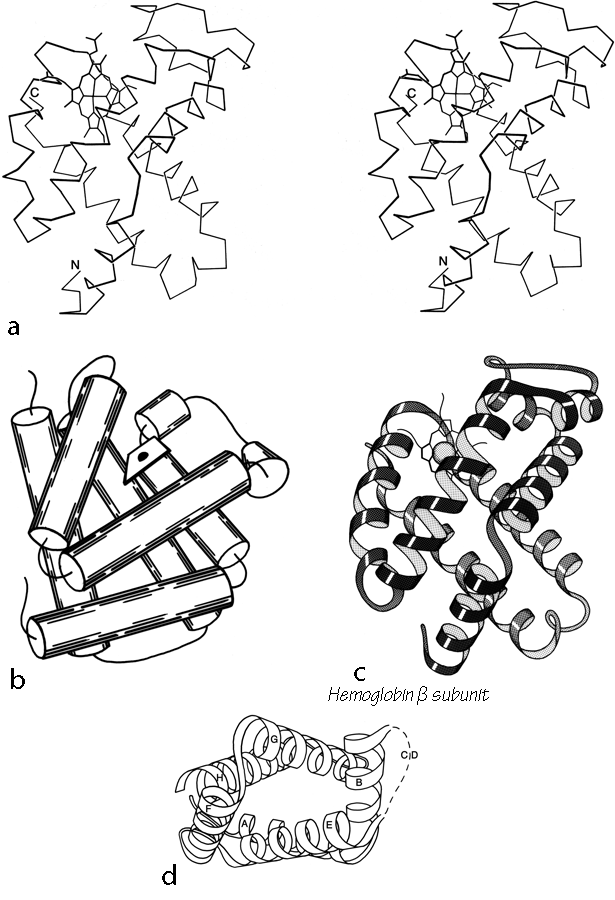
These four structures then form the second major subgrouping of antiparallel α domains, which we will call Greek key helix bundles (see Fig. 73). The helix elements lie on an approximate cylinder (see Fig. 89d for an end view), with O to 45° right-handed twist relative to the cylinder axis; they are connected with a Greek key topology which can have either a counterclockwise (globins) or a clockwise (thermolysin d2 and T4 lysozyme d2) swirl when viewed from the outside.
The remaining structures in this category (carp Ca-binding protein, egg lysozyme, citrate synthase, catalase d2, and ρ-hydroxybenzoate hydroxylase d3) are miscellaneous helical domains. However, there is good evidence from sequences and from functional resemblances (Kretsinger, 1976) that carp Ca-binding protein exemplifies a whole group of proteins that are constructed of "E-F hands" (see Section II,F) and that regulate or are regulated by changes in Ca2+ concentration. [This is now a large group typified by calmodulin. The “T-shaped” helix-turn-helix motifs of many transcription factors are superimposable onto E-F hands in the reverse sequence direction (Richardson).] (###REF)(###REF)(###) Citrate synthetase may be the first example of a group of larger helical domains with three layers. [The peroxidases (e.g. 2CYP) are examples, but the layers are usually not distinct.] Irregular helical structures with a moderate number of disulfides can be classified either here or as small SS-rich. We have classified egg lysozyme here (with only 4 disulfides in 129 residues), while phospholipase A2 (with 7 disulfides in 123 residues) is classified with the small SS-rich proteins.
[The most distinct new all-α folds are the cylindrical or crescent-shaped repeats of helix hairpins, both of which are exemplified by the two chains of farnesyl transferase.]
The largest grouping of structures contains domains organized around a parallel or mixed β sheet, the connections for which form structure (usually helical) protecting both sides of the sheet, with the helices also predominantly parallel to each other. Of course, each helix and its neighboring β strand are antiparallel to one another, but this structure category is called parallel α/β because both the β sheet interactions and the α-helix interactions are internally parallel. The parallel α/β category is the same as Levitt and Chothia's α/β proteins. Figures 75-78 show schematic drawings of this group of structures. It is interesting to note that there seems no a priori reason not to have parallel all-α structures or parallel all-β structures formed of two helix layers or of two parallel β sheets, yet such structures are not found. [There are now indeed examples of both these types, described in the annotations to sections III.B for helical horseshoes and III.D. for β-helices.] All of the domains with parallel organization have both a β sheet of at least four or five strands and at least three or four α-helices. Almost all have at least three layers.
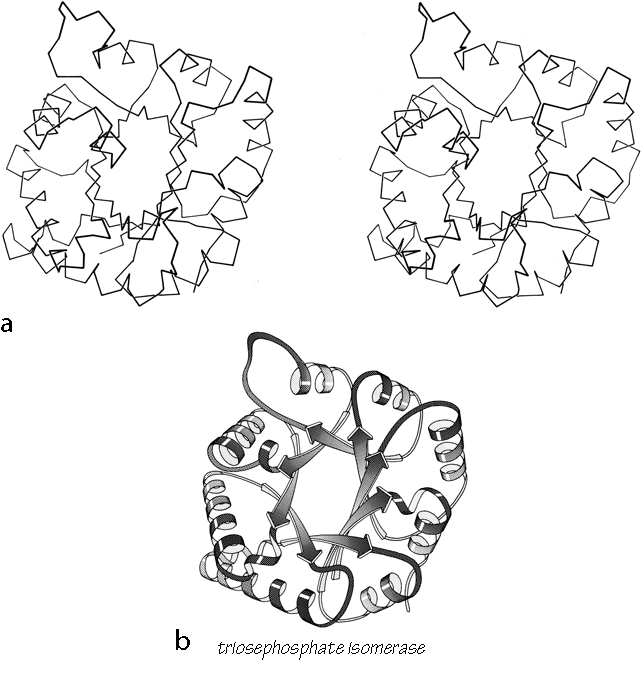
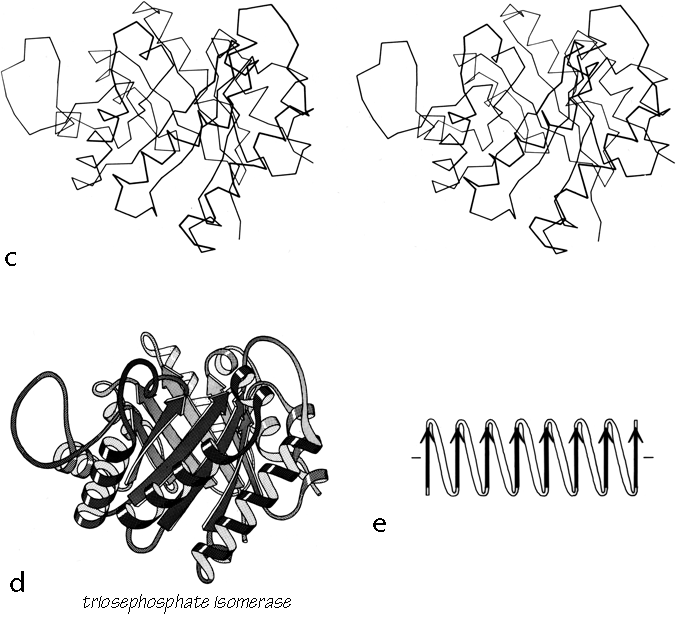
The first subgrouping under the parallel α/β category contains two of the largest but simplest domain structures that have yet been found. They are the eight-stranded parallel β barrels of triosephosphate isomerase (see Fig. 90) and pyruvate kinase domain d1, both of which are connected in +1x,+1x topology all the way around. (In structures with both β sheet and also several helices it is convenient to use just the β strands for designating the topology.) The connections are α-helices, which form a larger cylinder of parallel helices concentric with the β barrel. The structural elements of both α and β cylinders have a pronounced right-handed twist around the cylinder axis. Connections between the parallel β strands must lie on the outside of the barrel since the interior is filled by the packed hydrophobic side chains. If all of the crossover connections must be right-handed and no knots are allowed, then the chain must wind consistently around the barrel in one direction, and the +1x, +1x, +1x topology is not only the simplest but essentially the only possible topology for such a structure (Richardson, 1977), since all other topologies are knotted and unfoldable. We call this structure the singly wound parallel β barrel, since successive crossover connections are wound on the barrel progressing in a single direction with no reversal or backtracking. Figure 91a is a highly schematized representation of the "singly wound" structure, viewed from one end of the barrel.
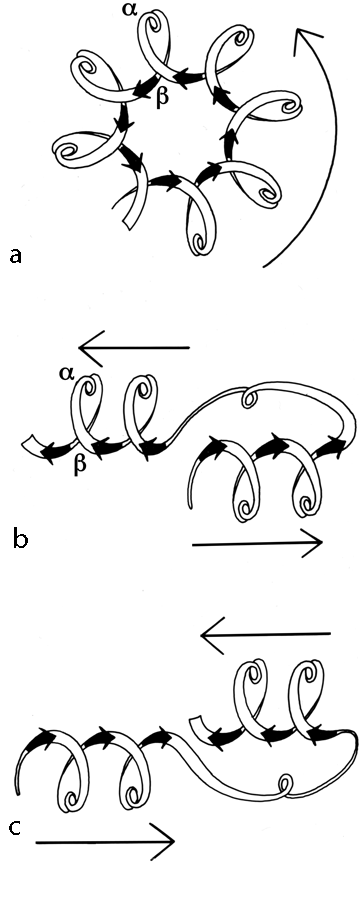
The largest subgrouping within the parallel α/β category contains structures with a central twisted wall of parallel or mixed β sheet, protected on both sides by its crossover connections (most of which are helical). This is called the doubly wound parallel β sheet, because with right-handed crossovers the simplest way of protecting both sides of the sheet is to start near the middle and wind toward one edge, then return to the middle and wind to the other edge. Figure 91b is a highly schematized representation of the "doubly wound" structure, viewed from one end of the sheet (compare with Fig. 91a). The singly wound barrel has four major layers of backbone structure and the doubly wound sheet has three major layers (with two separate hydrophobic cores); most other domain structures have only two major backbone layers with a single hydrophobic core, and are on the average considerably smaller.
The doubly wound structures were first recognized as a category by Rossmann in comparing flavodoxin with lactate dehydrogenase d1. As more and more protein structures were solved which fell into this category, the relationships between them have been described and debated at considerable length. The initial descriptions were in terms of the β—α—β—α—β unit as a supersecondary structure (Rao and Rossmann, 1973). Quite soon the emphasis shifted to the functional properties of the nucleotide-binding site which most of them share, and to the probable evolutionary relationships between these "nucleotide-binding domains" (Schulz and Schirmer, 1974c; Rossmann et al., 1974). By now the consensus appears to be that some of the most similar of these structures must certainly be related to each other, while at least some of the most dissimilar examples surely cannot be related (Rossmann and Argos, 1976; Matthews et al., 1977; Levine et al., 1978; McLachlan, 1979a).
We will group these domains into five gradually loosening levels of topological similarity, without attempting to make any definite decision as to where the dividing line lies between divergent and convergent examples.
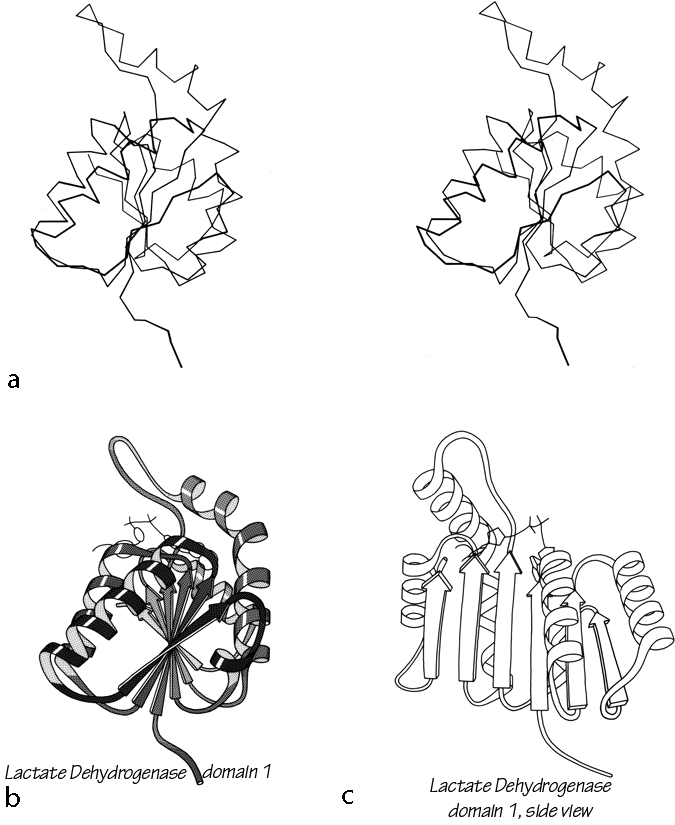
Figure 92 shows stereo and schematic drawings of lactate dehydrogenase domain 1, which is the classic example of a nucleotide binding domain or doubly wound sheet.

The darkest inner box in Fig. 93 includes those "classic" doubly wound parallel sheets that exactly match the topology of lactate dehydrogenase d1. Phosphorylase domain 2 is a five-layer structure in which the central three layers are a classic doubly wound sheet and the outer helical layers are formed by the two ends of the chain. The next box includes examples in which deleting one or two strands either at an end of the chain or at an edge of the β sheet will produce a five- or six-strand doubly wound sheet, while in the next box such deletions yield four doubly wound strands. In the outermost solid box it is necessary to omit strands interior to both the sheet and the sequence in order to get four strands of doubly wound β sheet. The structures inside the dotted box can yield no more than three such strands and cannot really be described as doubly wound, they share with the rest of this large subgrouping only the general organization of a central wall of parallel or mixed β sheet protected on both sides by its connections (see Fig. 94 for carboxypeptidase as an example). As one progresses outward from the classic to the most peripheral cases, the number of antiparallel strand pairs mixed in with the parallel gradually increases. Aside from the "classic" examples in the inner box, there are several other exact duplicates of doubly wound topologies between different proteins: phosphorylase d1 versus glyceraldehyde-phosphate dehydrogenase d1; aspartate transcarbamylase catalytic d1 versus rhodanese d1,d2; catalase d3 versus flavodoxin; and ρ-hydroxybenzoate hydroxylase d1 versus glutathione reductase d1.
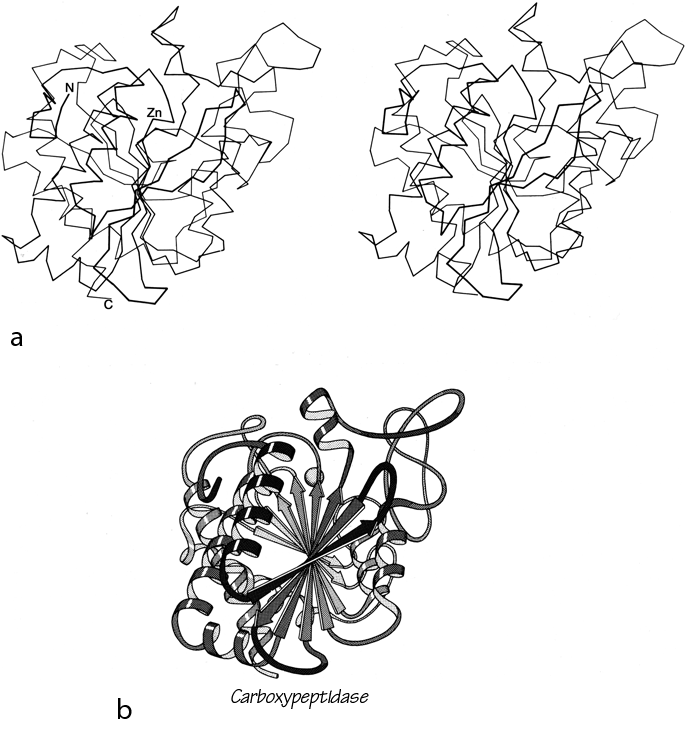
As one progresses from classic to peripheral doubly wound sheets, the number of domains that bind nucleotides also decreases. A favorable site for binding dinucleotides (or in a few cases, mononucleotides) is associated with this general category of structure and to a large extent with the doubly wound topology. The dinucleotides are all bound in approximately equivalent positions at the C-terminal end of the β sheet strands, within one strand of the central position where the winding switches direction (see Fig. 91b). [The importance of these "topological switch points" for active sites in doubly-wound domains is well explained in Branden and Tooze (1991).] Nucleotides are also bound at the C-termini of β strands in the singly wound barrels. In most of these cases, each nucleotide is associated with a "mononucleotide-binding fold" of three β strands and two helices with +1x, +1x topology; combination of two of these folds around a local 2-fold axis produces the classic doubly wound sheet. In some cases, however (such as hexokinase or dihydrofolate reductase), the topology is quite significantly different. Also there seems to be another quite different type of nucleotide-binding site such as the active site in staphylococcal nuclease (Arnone et al., 1971) or the AMP site in phosphorylase (Sygusch et al., 1977); both of these sites rely mainly on arginines for binding the nucleotide phosphates.
One quite surprising and intriguing feature of this group of structures is that it contains extremely few examples of the "reverse doubly wound" topology (see Fig. 91c), a different but equally plausible pattern related to the doubly wound sheet by reversing the N- to C-terminal direction of the chain or by switching relative positions of the two halves of the β sheet. Of the four reverse examples (found in PGK d2, GPDH d1, glucosephosphate isomerase d1, and phosphorylase d1) none forms a nucleotide-binding site, all belong to a sheet that also has a normal doubly wound section, and none includes more than four strands. Those cases do demonstrate that such a topology is stable and can fold, but there must be some strong reason why it is so rare. Some simple explanations of this regularity would be either that most of the nucleotide-binding domains are related, or that they must fold strictly from the N-terminus, or that the requirements for forming a nucleotide-binding site are restrictive enough to constrain the usual doubly wound topology. None of these explanations is completely satisfying, however, because a number of domains are known that cannot fold strictly from the N-terminus, because the relative placement of features forming the nucleotide sites is only rather approximate (e.g., see D. A. Matthews et al., 1979), and because the rearrangement necessary to produce a reverse doubly wound sheet seems much less drastic than many of the rearrangements that must be proposed if all of these proteins are related.
The next major grouping consists of domains that are organized around an antiparallel β sheet. They are as numerous as the parallel α/β structures, and their topology and classification have been discussed before (see Levitt and Chothia, 1976; Sternberg and Thornton, 1977a,b; Richardson, 1977). This category is the most varied in terms of size and organizational patterns. Figures 79 through 84 show backbone schematics for the antiparallel β domains, grouped into subcategories.
Most of the antiparallel β domains have their β sheets wrapped around into a cylinder, or barrel, shape. None of the antiparallel barrels has as symmetrical or as continuously hydrogen-bonded a cylindrical sheet as the singly wound parallel β barrels of triosephosphate isomerase and pyruvate kinase d1; however, antiparallel barrels are very much more common. Because of gaps in the hydrogen bonding, some of these structures have been described as two β sheets facing each other (e.g., Schiffer et al., 1973; Blake et al., 1978; Harrison et al., 1978). Our reasons for treating them all as barrels are that the gap positions are sometimes different in domains that are probably related, and that the barrel description yields very much simpler and more unified topologies.
Barrels seem to prefer pure parallel or antiparallel β structure even more strongly than does β sheet in general. All the known singly wound barrels are pure parallel. An antiparallel barrel with an odd number of strands is constrained to have one parallel interaction, but no other parallel strand pairs occurs within antiparallel barrels except in the acid proteases. [ Of course, there are now more absolute numbers of such exceptions, but they are still quite rare. ] Also, even-stranded barrels are much more common than odd-stranded ones.
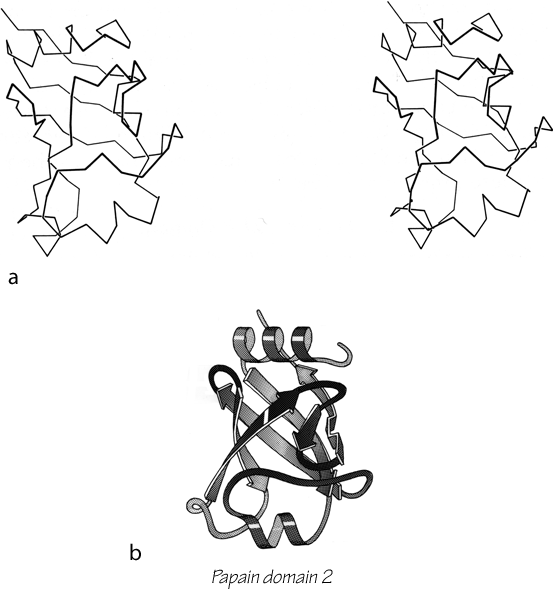
The first type of antiparallel β barrel, in analogy with the first type of helix bundle, has simple up and down +1, +1, +1 connections all around. Although it is relatively unusual for a barrel to be composed entirely of up-and-down strands, many of the larger barrels and sheets have four- to six-stranded sections of simple up-and-down topology embedded within them. There are three examples of pure up-and-down β barrels: soybean trypsin inhibitor, papain d1, and catalase d1. [ Two interesting later examples are retinol-binding protein (1KT7) and the large, membrane-spanning, 16-strand or more up-and-down barrel of porins (e.g. 1HXX). ] Figure 95 shows a stereo and a schematic drawing of papain d1. Soybean trypsin inhibitor has long excursions at the ends of three of the β strand pairs, forming separate, twisted β ribbons; there is a strong internal 3-fold symmetry which includes these ribbons as well as the strand pairs in the barrel (McLachlan, 1979C). Catalase d1 is an eight-stranded up-and-down barrel with less extreme loop excursions. Rubredoxin could be considered as a very irregular and incomplete up-and-down barrel in which β-type hydrogen bonds are formed between only about half of the strand pairs (see Fig. 76). It is very small and compact, and is presumably stabilized partly by the network of Cys ligands to the iron; therefore we have placed it in the small metal-rich category.
Soybean trypsin inhibitor, papain d1, and rubredoxin have identical topologies: six strands of +1, +1, +1, . . . proceeding to the left around the barrel if the chain termini are at the bottom. However handedness is not nearly as meaningful a property for up-and-down topologies as it is for Greek keys, since up-and-down handedness can change on addition or defection of a single strand.
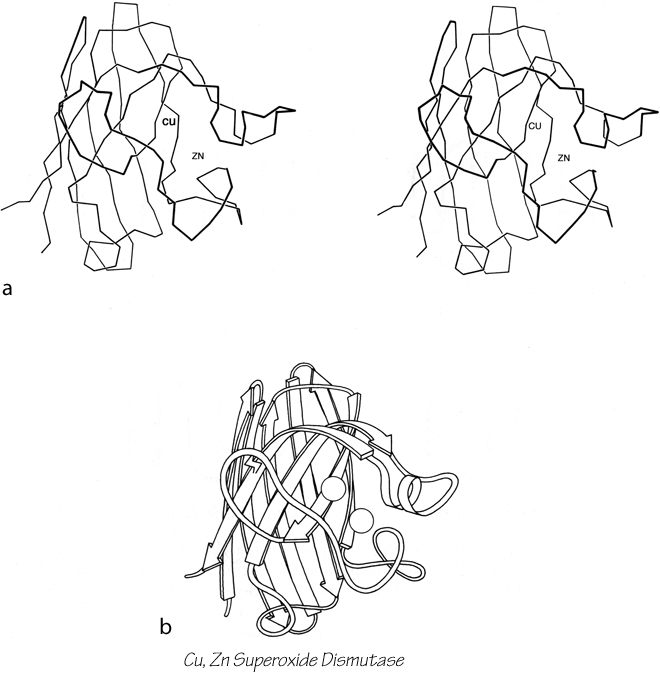
The commonest subgroup of antiparallel β barrel structures has a Greek key topology, with -3, +1, +1, -3 connections or a close variant. The first Greek key barrel structures were compared in Richardson et al. (1976), and they and the up-and-down barrels were described as categories in Richardson (1977). Figure 96 illustrates Cu,Zn superoxide dismutase as an example of a Greek key β barrel. There are 13 Greek key barrels in our sample, and 12 of them (all except staphylococcal nuclease) have the same handedness: viewed from the outside, the Greek key pattern forms a counterclockwise swirl (see Fig. 97). The four barrels shown in Figure 81 have a more complicated "jellyroll" topology with an extra swirl in the Greek key (this pattern was also common on Greek vases); the "jellyroll" Greek key topologies are shown in Fig. 98. The jellyroll pattern is produced by having a pair of connections, rather than just one connection, crossing each end of the barrel.
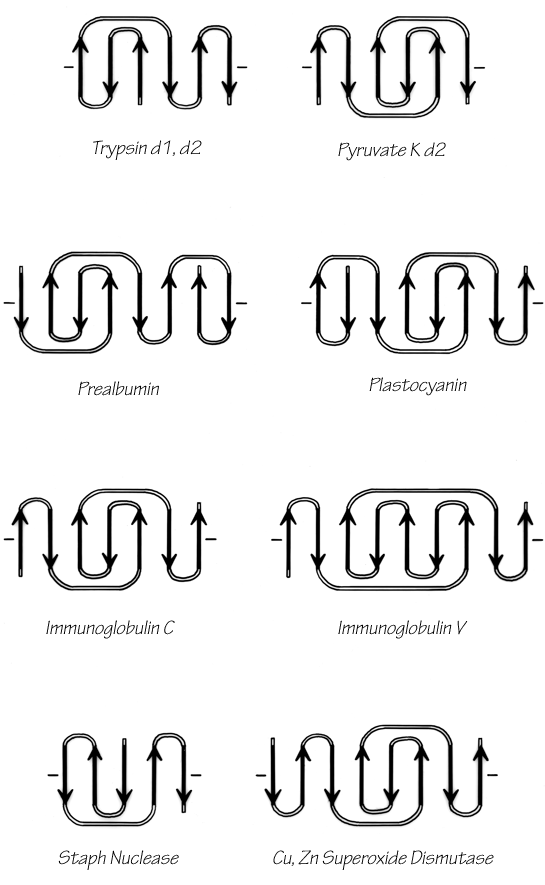
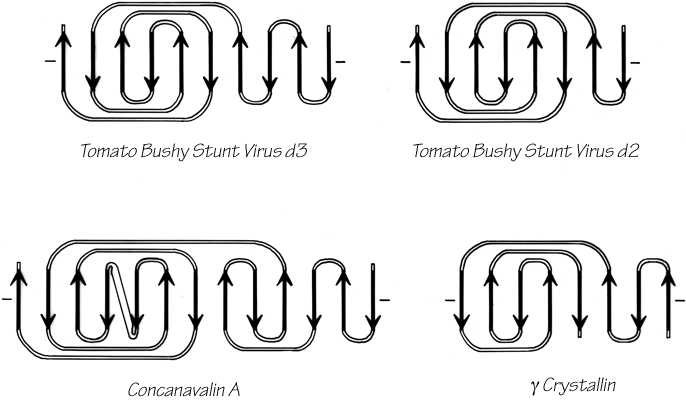
The Greek key barrels have between 5 and 13 strands, but in all cases they enclose approximately the same cross-sectional area (see Section II,B). The cross sections are somewhat elliptical, with more flattening the more strands there are. For 8- to 10-stranded barrels, it is noticeable that the direction of the long axis of the cross-section twists from one end of the barrel to the other by close to 90° (see Fig. 99)

± 3 connections are not particularly common outside of the barrels so that the prevalence of Greek key topologies is not due simply to chance combination of the connection types that make it up. There are two different ways of analyzing the Greek key which could perhaps explain both its frequent occurrence and its strongly preferred handedness. The first approach is to consider the stability of the final barrel, given its size, shape, and twist. Figure 99 shows that the Greek key pattern provides neat, efficient connections across the top and bottom of the barrel, lhydrogen bonding next to the ±1 connections. In tomato bushy stunt virus d3 there is actually some β-type hydrogen bonding between the +1, -3, and +5 connecting loops. In combination with the twist of the strands and of the barrel cross section, a counterclockwise Greek key (as shown) produces ±3 connections that are approximately perpendicular to each other on opposite ends of the barrel and that can both cross along a short axis of the cross section. A clockwise Greek key would place the ±3 connections in a weaker position approximately parallel to each other, and one of them would be along the long axis. This argument could not account for the handedness of the partial Greek keys with -3, +1, +1 topology (such as staphylococcal nuclease and chymotrypsin) where there is a ±3 connection at only one end of the barrel.
The other possible explanation hypothesizes an effect during the protein folding process, very similar to the one proposed to explain crossover handedness (see Section II,B). All Greek keys, even the "jellyrolls," necessarily have a folding point halfway along the chain from which two paired strands can be followed back next to each other as they curl around the structure. Given the prevalence of Greek key patterns in the known structures, it seems very likely that the polypeptide chain can fold up by first folding in half and forming a long, two-stranded β ribbon, and then curling up the ribbon to produce the further β sheet interactions. This sort of process is illustrated in Fig. 100. Since the initial ribbon would presumably have a strong right-handed twist (see Section II,B), it would impart a right-handed twist to the curling direction and always end up with a counterclockwise Greek key. Besides the β barrels, there are other pieces of protein structure that suggest this sort of process, such as the long β ribbons in lactate dehydrogenase d2 (see Fig. 74). This kind of folding hypothesis has been utilized by Ptitsyn and Finkelstein (1980) to obtain rather successful predictions of β strand contacts and topologies. [ Ray Salemme (1983) ref ?? explained the curling up of a long, 2-stranded ribbon by the fact that φ,ψ preferences are slightly different for β-hairpin residues between a narrow pair of H-bonds and those between a wide pair, such that the former prefer the concave side of a curl and tend to end up on the inside. ]
Partial, multiple, and other barrels have been grouped together as another subgroup within the antiparallel β category (see Fig. 82). Ribonuclease contains a four-stranded antiparallel β sheet that looks 1ike a five-stranded barrel with one strand missing. Alcohol dehydrogenase d1 includes a five-stranded antiparallel barrel (with a topology of +1, +3x, -2, +1) and another partial five-stranded barrel. Back-to-back β barrels that share one wall occur in the variable half of immunoglobulin Fab structures (except for Rhe: see Wang et al., 1979), where VL and VH are each antiparallel β barrels and the contact between them forms an even more regular eight-stranded barrel with four strands contributed from each domain (see Fig. 101). The three barrels pack against each other with a right-handed superhelical twist, and the angle between the axes of adjoining barrels is the same as the angle between opposite strands in one of the barrels. The two domains of the acid proteases have complicated, very similar mixed β sheets that could be described either as a six-stranded barrel with side sheets or as several interlocking β sheets. When more examples are available, it will probably be possible to find patterns to the ways in which small subsidiary β sheets can interlock into the edges of larger sheets (such as in the acid proteases or thermolysin d1), but for now no attempt has been made to classify them.
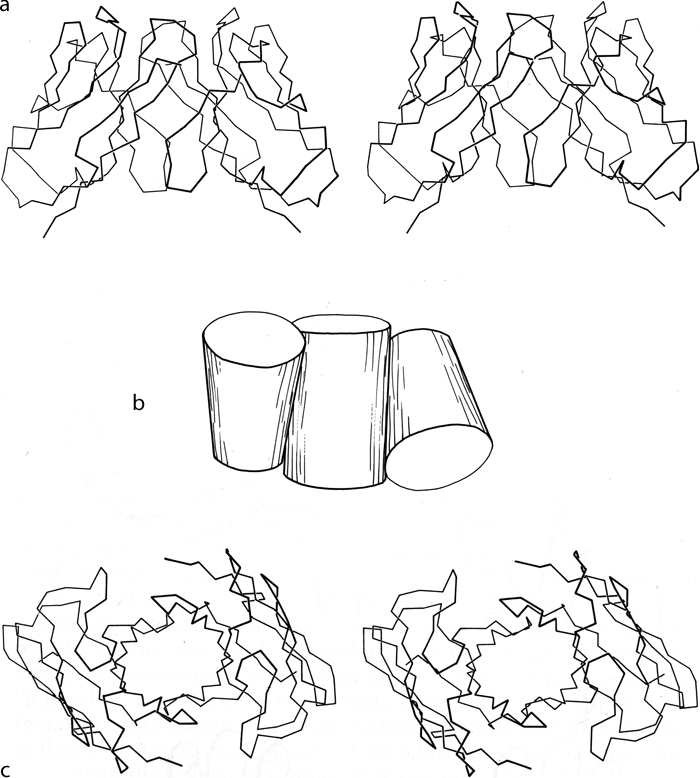
The next subcategory of antiparallel β domains each has a single, more or less twisted β sheet, either pure antiparallel or predominantly so, but not closing around to form a barrel. They are shown in Fig. 83, and Fig. 102 shows glyceraldehyde-phosphate dehydrogenase as an example. Their common feature is a layer of helices and loops which covers only one side of the sheet, so that they are two-layer structures. Many β barrels have been described as "sandwiches," with two slices of β sheet "bread" and a "filling" of hydrophobic side chains; based on that analogy these structures would be "open-face sandwiches," with a single slice of β sheet "bread" and a "topping" of helices and loops. The open-face β sandwiches could rival a Danish buffet for variety on a theme: they range from 3 to 15 strands, with a wide assortment of topologies, curvatures, and placement of helices and loops. Bacterio-chlorophyll protein, the largest of them, encloses between the β sheet layer and the helical layer a core of seven bacteriochlorophyll molecules, tightly packed in an orderly but quite asymmetrical array.
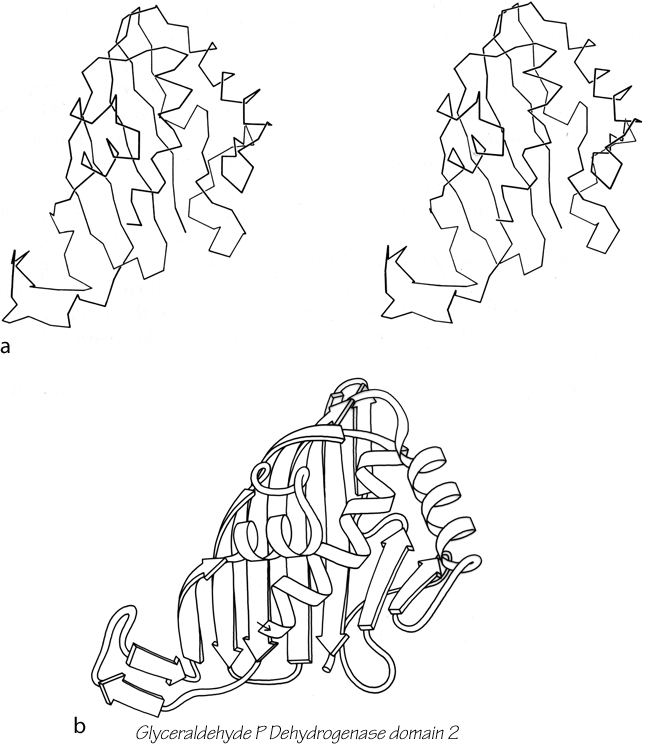
The remaining three antiparallel β structures form a miscellaneous category (see Fig. 84). Lactate dehydrogenase d2 and gene 5 protein each has several two-stranded antiparallel β ribbons, but they do not coalesce into any readily described overall pattern. The N-terminal domain of tomato bushy stunt virus protein has a unique β structure in which equivalent pieces of chain from three different subunits wrap around a 3-fold axis to form what has been called a "β annulus" (Harrison et al., 1978). Each of the three chains contributes a short strand segment to each of three three-stranded, interlocking β sheets. This "domain" provides one of the subunit contacts that hold the virus shell together. However, only one-third of the 180 subunits contribute to the β annuli; for the other quasi-equivalent subunits, the N-terminal part of the chain is disordered with respect to the virus shell.
[ Many new variants of all-β structures have been seen more recently, including two entirely unprecedented fold types. The β-propeller is made up of 4, 5, 6, 7, or 8 units, each a small 4-stranded up-and-down β sheet, arranged radially like propeller or rotor blades around a center of approximate symmetry (e.g., 1TBG G protein β subunit). The β-helix is a parallel all-β structure, which winds around in a shallow spiral forming 3 parallel β-sheets in a triangular cylinder. It comes in both righthanded (1O88 pectate lyase C) and lefthanded (1LXA Lpx A acyltransferase) forms, with different cross-sectional shapes, and is the only handed protein structure that commonly occurs in either handedness. ]
The last major category (shown in Figs. 85 and 86) consists of small (usually less than 100 residues) domains whose structures seem to be strongly influenced by their high content either of disulfide bonds (S) or of metal ligands (M). These S—M proteins often look like distorted versions of other, more regular, proteins. The disulfide-rich ones include many toxin and enzyme-inhibitor structures. For most of the disulfide-rich proteins it is known experimentally that they are completely unstable if the disulfides are broken (in contrast to larger disulfide-containing proteins, for which disulfides merely provide additional stabilization for an already-determined structure). Figure 103 shows pancreatic trypsin inhibitor as an example of a disulfide rich protein, and Fig. 104 shows cytochrome c as an example of a metal-rich protein. Most of the S—M proteins are single-domain and monomeric: only wheat germ agglutinin has multiple domains, and only insulin has multiple subunits in the molecule.
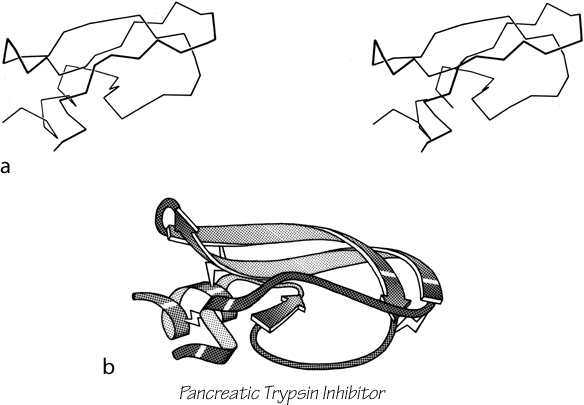
The only subgroup of similar structures within the S—M proteins is the toxin-agglutinin folds of the snake neurotoxins and the domains of wheat germ agglutinin (see Fig. 85). They are made up of extended-chain loops with an almost identical topology of -1, +3, -1, +2x rather like a series of half-hitch knots (the β structure is extremely minimal in wheat germ agglutinin) strongly linked by a core of four disulfides, three of which are equivalent (see Drenth et al., 1980). High-potential iron protein and ferredoxin share a local loop structure that binds the iron-sulfur cluster, but otherwise are different.
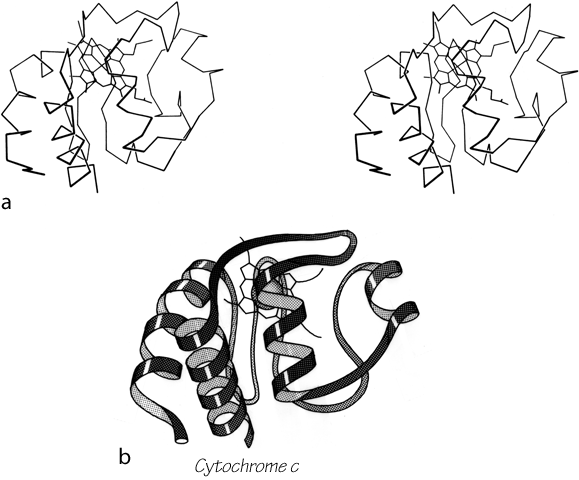
Most of the metal-rich proteins form approximately cylindrical two-layer structures with either an up and down (rubredoxin, cytochrome c) or a Greek key (ferredoxin) topology, but in which the elements forming the cylinder are a mixture of helices, β strands, and more or less extended portions of the backbone. Cytochrome c3 is perhaps the ultimate example of an S—M protein, with four hemes in just over a hundred residues, and essentially no secondary structure at all except for one helix.
One way of considering these proteins is as distorted versions of the other structural types. Most S—M proteins can fairly clearly be grouped as either distorted helix clusters (phospholipase, cytochrome c, cytochrome b5), distorted β barrels (rubredoxin, high-potential iron protein), or distorted open-face sandwiches (erabutoxin, wheat germ agglutinin, pancreatic trypsin inhibitor, or ferredoxin). Figure 105 shows an example of each of these relationships. In fact, one reasonable taxonomy would do away with this fourth major category altogether and place all the S—M proteins as irregular examples of either an α or a β category. We have not chosen that approach, however, because several of the structures (crambin, insulin, and cytochrome c3) are rather difficult to place in one of the other categories, and also because these small proteins influenced by nonpolypeptide interactions appear to share important features, especially in terms of the probable complexity of their folding process (see Section IV,C).
Another suggestive fact is that there are no small, irregular structures related to the parallel α/β category. Perhaps this reflects the fact that domains organized around parallel β sheet are necessarily fairly large and seem to be dependent on large, buried, and quite regular β structure for their stability. There are in fact no hemes (in spite of all the helices) or iron-sulfur clusters in parallel α/β proteins, and no disulfides except for the single active-site disulfides of thioredoxin, glutathione peroxidase, and glutathione reductase.
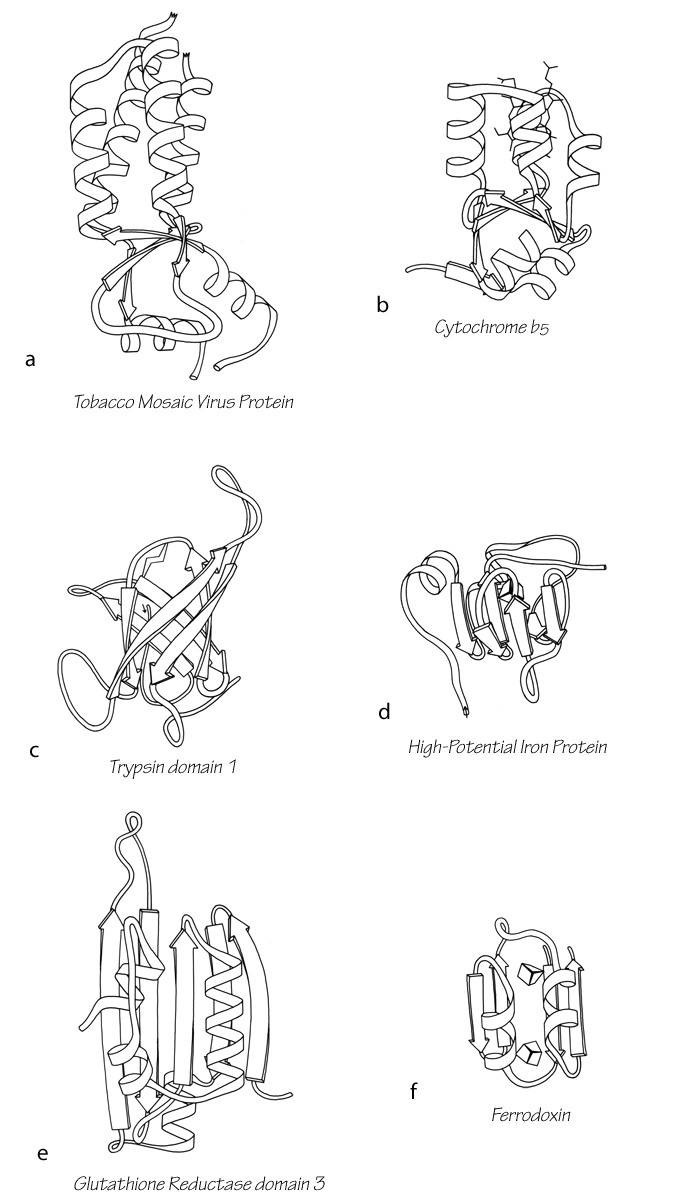
Aside from the direct techniques of X-ray or electron diffraction, the major possible routes to knowledge of three-dimensional protein structure are prediction from the amino acid sequence and analysis of spectroscopic measurements such as circular dichroism, laser Raman spectroscopy, and nuclear magnetic resonance. With the large data base now available of known three-dimensional protein structures, all of these approaches are making considerable progress, and it seems possible that within a few years some combination of noncrystallographic techniques may be capable of correctly determining new protein structures. Because the problem is inherently quite difficult, it will undoubtedly be essential to make the best possible use of all hints available from the known structures.
The most important general point to be emphasized is that it is now both possible and essential to frame and test hypotheses about exactly which structural features a given technique is really measuring or predicting. Structure surveys like the current one can help in choosing proteins that will provide critical tests of such hypotheses both by locating proteins that vary most in the parameters under consideration, and also in helping to control for the effects of differences in other major structural parameters. It seems inherently unlikely that any spectroscopic feature is a direct measure of percentage α,β turn, and coil as defined in any of the usual ways. But since such percentages are certainly not the only useful way to describe protein structure, it should be fruitful to combine theoretical analysis (where possible) with careful empirical tests in order to determine the set of descriptions most applicable for a given technique. Let us consider several examples of what could be attempted with this approach.
Methods for predicting secondary structure from amino acid sequence could presumably benefit from considering parallel and antiparallel β sheet separately, since the two types have rather different single and pairwise residue preferences. An overall classification scheme could help in choosing a large and characteristic sample. However, there is the difficulty of dealing with mixed β sheets. For a given set of parameters that successfully distinguished pure parallel from pure antiparallel sheets, it would be possible for instance to test whether the characteristics of strands in mixed β sheets depended mainly on their local hydrogen-bonding type or depended mainly on whether the overall sheet organization was "antiparallel β" type or "parallel α/β" type (for example, prealbumin is an "antiparallel" mixed sheet and carboxypeptidase is a "parallel" mixed sheet). This sort of question should be asked also for the amide I bands in infrared (see Miyazawa and Blout, 1961; Chirgadze and Nevskaya, 1976) and Raman spectra (see Krimm and Abe, 1972; Yu et al., 1975) that are thought to be sensitive to the differences between parallel and antiparallel β sheet. It would be especially useful if it turned out that some features were sensitive to local and some to overall structure. In general, the parallel α/β structures have been grossly underrepresented in spectroscopic studies of protein conformation, because they do not occur in the small proteins that made up most of the early X-ray structure determinations. Now that α/β proteins have been shown to be extremely common, this sampling bias can be corrected.
The C—S and S—S stretch vibrations of disulfides (Edsall et al., 1950) can be observed in the Raman spectra of proteins, but their interpretation is still somewhat controversial (see, for example, Klis and Siemion, 1978; Spiro and Gaber, 1977). Using series of model compounds, Van Wart et al. (1973) have related S—S stretch frequency to the χ3 (Cβ—S—S—Cβ) dihedral angle, while Sugeta et al. (1972, 1973) have related the S—S frequency to the χ2 (Cα—Cβ—S—S) dihedral angle and C—S stretch frequency to χ1 angle; these latter correlations have been further modified by Van Wart and Scheraga (1976). The relationship of spectrum to conformation seems to be quite complex in proteins, where constraints at either end of the disulfide would tend to increase coupling between the modes. The S—S stretch may be sensitive to the relative sign as well as the absolute value of χ2, and therefore may reflect the difference between the spiral and the hook conformations (see Section II,E). It should be possible to determine characteristic spectra for the three common disulfide conformations found in proteins (the left-handed spiral, the right-handed hook, and the extended form in immunoglobulins) by choosing accurately refined proteins with a single or a dominant disulfide conformation (e.g., immunoglobulins, carboxypeptidase, egg lysozyme, and pancreatic trypsin inhibitor).
Very low-frequency vibrations have been observed in proteins (e.g., Brown et al. 1972; Genzel et al., 1976), which must involve concerted motion of rather large portions of the structure. By choosing a suitable set of proteins to measure (preferably in solution), it should be possible to decide approximately what structural modes are involved. Candidates include helix torsion, coupled changes of peptide orientation in β strands, and perhaps relative motions of entire domains or subunits. These hypotheses should be tested, because the low frequency vibrations probably reflect large-scale structural properties that would be very useful to know.
In using circular dichroism to estimate percentages of the various secondary structures in a protein (e.g., Saxena and Wetlaufer, 1971; Grosse et al., 1974), helix can be judged more reliably than other features, as is usually true for almost any method including prediction (e.g., Maxfield and Scheraga, 1976). This is presumably because α-helices are relatively uniform in both local and longer range patterns, while β structure is widely variable in hydrogen-bonding pattern, regularity, twist, exposure, and overall shape. There is at least a real possibility that differences in shape and organization of β structure are reflected in the circular dichroism spectrum; that possibility should be tested, because it would be even more useful to be able to categorize a structure as a doubly wound sheet or an antiparallel β barrel than to say it had 35% β structure, even supposing that we could reliably do the latter.
Successful examples of the sort of correlations postulated above would add additional independent pieces of information for use in a combined strategy of noncrystallographic protein structure determination. Empirical regularities such as the handedness of crossover connections (see Section II,B) can help in narrowing down the possibilities. Another need is to decide whether, and at what point, a protein is divided into domains. The more tenuously connected domain pairs can often be recognized by such techniques as electron microscopy, viscosity, low-angle scattering, or proteolysis, and it might prove possible to recognize domain-connection regions in the sequence. Knowledge of a set of common overall structure types (such as the major subgroupings in our classification scheme) can provide prototypes with which to match the distribution of predicted secondary structures and the characteristics suggested by various spectroscopic measures. For a given protein, combination of all these methods in an overall strategy that can deal with their probabilistic nature and disparate information content may some day be able to produce a fairly small number of alternative structures, one of which (by some process such as energy minimization) would converge to what could be recognized as the correct native structure.
Even an infallible method of structure prediction would not make protein crystallography obsolete; detailed prior knowledge of the globin structure has not removed the necessity or interest of high resolution X-ray structures for other species, mutants, and ligand forms of hemoglobin. What it would do is to take away a great deal of the fun and excitement of discovering new structures by protein crystallography; but that is not too large a price for the kind of increased understanding that is likely to accompany even the most ad hoc of successful structure prediction methods.
One important reason for classifying proteins is simply to make the structures more memorable. The system proposed above can help to do that, especially for those cases which fall into one of the more narrowly defined subgroups. However, we also want to know to what extent this classification is a true taxonomy: that is, whether or not it expresses underlying genetic relationships. In addition, among so many structure examples, almost any major rule governing either protein evolution or protein folding would have predictable statistical consequences on the pattern of structural resemblances to be expected. Therefore, it is worthwhile examining the distribution of features that is actually found, because it may suggest various conclusions about how proteins evolve and fold.
Similar domain pairs |
Different domain pairs |
| Phosphorylase d1, d2 | Papain d1, d2 |
| Phosphoglycerate kinase d1, d2 | Tyrosyl-tRNA synthetase d1, d2 |
| Aspartate carbomoyltransferase catalytic d1, d2 | Thermolysin d1, d2 |
| Arabinose-binding protein d1, d2 | T4 phage lysozyme d1, d2 |
| Phosphofructokinase d1, d2 | Glucosephosphate isomerase d1, d2 |
| Rhodanese d1, d2 | Pyruvate kinase d1, d2 |
| Hexokinase d1, d2 | Pyruvate kinase d2, d3 |
| Glutathione reductase d1, d2 | Lactate dehydrogenase d1, d2 |
| Tomato bushy stunt virus d2, d3 | Alcohol dehydrogenase d1, d2 |
| Chymotrypsin d1, d2 | Glyceraldehyde-phosphate dehydrogenase d1, d2 |
| γ-Crystallin d1, d2 | Glutathione reductase d2, d3 |
| Immunoglobulin C, V | Influenza virus hemagglutinin HA1, HA2 |
| Immunoglobulin C1, C2 | p-Hydroxybenzoate hydroxylase (4-hydroxybenzoate 4-monooxygenase) d1, d2 |
| Immunoglobulin C2, C3 | p-Hydroxybenzoate hydroxylase d2, d3 |
| Acid protease d1, d2 | Catalase d1, d2 |
| Wheat germ agglutinin d1, d2 | Catalase d2, d3 |
| Wheat germ agglutinin d1 and d2, d3 and d4 | |
a For proteins with more than two domains, each potential duplication is listed separately: e.g., a minimum of two duplications would be necessary to produce either a three-domain or a four-domain structure. Members of the pairs in the left-hand column both fall within the same structural subcategory and have fairly similar topologies; such pairs are perhaps the result of internal gene duplications. Members of pairs in the right-hand column almost all fall into different major categories of tertiary structure (e.g., one all-helical and one antiparallel β); presumably they could not have been produced by internal gene duplication. | |
One significant feature evident in the known structures is the frequency with which domain pairs within a given protein are found to match each other closely in structure. It is known from amino acid sequences (e.g., Dayhoff and Barker, 1972) that internal gene duplication can occur in proteins. For recent or highly conserved duplications with closely related sequences the duplication event can be conclusively demonstrated. However, study of sequences cannot tell us how widespread and frequent gene duplication has been in the evolution of proteins because it cannot detect old duplications whose sequences have had time to diverge beyond recognizable homology. There are 26 multidomain proteins in our sample, which would have required the introduction of new domains 35 different times; they are listed in Table II. In slightly over half (17) of those cases, the structure of the new and old domains is basically the same (Fig. 106 shows the two domains of rhodanese as an example); in two cases (cytochrome c peroxidase and aspartate transcarbamylase regulatory chain) the level of similarity is ambiguous; while in the other 16 cases the structures are totally different and presumably could not be the result of internal gene duplication (e.g., Fig. 107). Many of the 17 similar cases involve rather unusual structures, such as the complex mixed sheets of the acid proteases, the five-layer domains of phosphorylase or the mixed doubly wound sheets of hexokinase (Fig. 108).

In only 4 of the 17 similar domain pairs is it possible to find a domain in some other protein that matches the structure of one of the pair as well as they match each other (two of those four cases are classic-topology doubly wound sheets). Only for the immunoglobulins (and probably for wheat germ agglutinin, if its sequence were known) is there any significant sequence homology detectable between the similar domain pairs. Purely by chance one would expect vaguely similar structures (within the same major subgroup) perhaps one time out of ten, and detailed resemblance of relatively unusual structures only about one time in 50 or 100. It is unlikely therefore, that more than one or two of the 17 similar domain pairs happened by chance. Only one or two of the pairs could be the products of convergent evolution, because in the other cases the two domains of the pair have quite different functions. Therefore it seems fairly certain that almost all of the similar domain pairs are indeed the result of internal gene duplications. We are left then with the rather interesting conclusion that about half the time multiple domains are produced by gene duplication.
It is also possible that the large and relatively complex domain structures we find today were initially produced by gene duplication from smaller substructures; many of these cases have been analyzed by McLachlan (e.g., McLachlan et al., 1980; McLachlan, 1979b,c). There is very strong evidence from sequence as well as structure that this occurred in ferredoxin (Adman et al., 1973) and probably in the carp calcium-binding protein (Kretsinger, 1972). These substructures would not have been very stable by themselves, but they could perhaps have survived under less rigorously competitive conditions by associating as identical subunits. (There must have been a stage early in the evolution of life when there were few proteases to degrade a temporarily unfolded protein; also, marginally stable proteins become a selective disadvantage only when other organisms develop more stable ones.) It is very difficult to tell from the present structures whether or not this process commonly occurred. Most such possible substructures are so simple that they are very likely to occur often no matter how the domains originated, which means that the internal symmetry of the doubly wound, singly wound, up-and-down α, and up-and-down β structures does not prove that they originated by duplication Also, such duplications would have to have been extremely ancient genetic events and there could have been much alteration since then, so the fact that most other structures show no internal symmetries does not prove that they did not originate by such a process. For example, the uteroglobin subunit (see Fig. 88) is not internally symmetric because its fourth helix exchanges position with the one on the other subunit of the dimer; it is very unclear, however, that uteroglobin is any less likely to be the product of an internal duplication than, for instance, myohemerythrin.
The next evolutionary question is how many of the different proteins are related to one another. To what extent are the various proteins in one of the structure subgroups all related? To what extent are proteins within a functional category related? In the end, this question comes down to asking how difficult it is to originate completely new proteins: are there very many, or relatively few, independent evolutionary lines among the proteins? At one extreme we would expect to see a fairly small number of distinguishable general structure types, and all members of one functional category of proteins would usually be found within the same structural class. If there were improbable structures represented, as there might well be, there would be only a few different such structure types but each would include several similar protein examples. At the other extreme, we would expect a fairly random distribution of functional types within the various structural categories, with many different improbable structures represented by only a single example of each. Neither of these extreme situations quite applies to the observed distribution of protein structures, but in general it conforms much more closely to the multiple-origin, random model. The simplest, most probable structure types are extremely common, while the more peculiar, complicated patterns each show up only once or twice.
In a very broad overview of the structural categories one can state several statistical correlations with type of function. Hemes are almost always bound by helices, but never in parallel α/β structures. Relatively complex enzymatic functions, especially those involving allosteric control, are occasionally antiparallel β but most often parallel α/β. Binding and receptor proteins are most often antiparallel β, while the proteins that bind in those receptor sites (i.e., hormones, toxins, and enzyme inhibitors) are most apt to be small disulfide-rich structures. However, there are exceptions to all of the above generalizations (such as cytochrome c3 as a nonhelical heme protein or citrate synthase as a helical enzyme), and when one focuses on the really significant level of detail within the active site then the correlation with overall tertiary structure disappears altogether. For almost all of the dozen identifiable groups of functionally similar proteins that are represented by at least two known protein structures, there are at least two and sometimes four or five totally different tertiary-structure types which share that function, as shown in Table III. Probably the most dramatic case is the proteolytic enzymes: although the trypsin-like serine proteases form a structurally related group, the proteases as a whole are represented by six widely different structures, including two textbook examples of convergent evolution: subtilisin versus the trypsin family and thermolysin versus carboxypeptidase. Even in the cases in which only two protein structures are known from a general functional category (such as lectins, nucleases, peroxidases, oxygen carriers, etc.) those two structures are quite different. It seems possible, then, that active sites are easier to alter or to redevelop independently than one would have thought, compared with the total time scale involved in the evolution of proteins.
|
|
|
aWithin each functional grouping, proteins known to be homologous are listed on a single line, and proteins that fall within the same tertiary-structure subcategory (for at least one of their domains) are bracketed. In spite of the detailed resemblances commonly found within active sites, the great majority of examples shown no similarity of overall structure.
|
|
The really puzzling fact, however, is that there is one glaring exception to the above pattern: the nucleotide-binding domains, especially the dehydrogenases and kinases. Within that functional group, and within the parallel α/β structures, the distribution is exactly what one would expect from the model in which large groups of proteins share an evolutionary origin (for at least one of their domains). Such a pattern could also be explained by especially stringent selective pressures, although there is no evidence at all that the requirements for nucleotide-binding sites are any more restrictive than for any other function (see Section III,C). Or the pattern could result from a combination of moderate selective pressure and some as-yet-unknown restrictions on folding within this general structure category. Whatever the explanation, it must somehow account for the fact that nucleotide-binding proteins (or, perhaps, enzymes in the glycolytic pathway) appear to be different in some fairly fundamental way from any other functional category sampled so far.
Another general approach that has commonly been taken to the problem of evaluating relationships between proteins is calculation of the minimum root-mean-square difference between superimposed a-carbon positions for the similar parts of the structures. This was initially done by Rossmann (Rao and Rossmann, 1973) to compare nucleotide-binding domains and other structures (e.g., Rossmann and Argos, 1976). These Cα comparisons in general corroborate and quantitate similarities found by inspection, and sometimes have uncovered relationships no one had previously noticed. Considerable progress has been made recently on ways of evaluating the significance level of Cα superpositions (Remington and Matthews, 1980; McLachlan, 1979b; Schulz, 1980). Two logically distinct problems are involved: the first problem is evaluating the significance of a given similarity relative to the probability of its "chance" or "random" occurrence, the second problem is estimating the likelihood that a given significant resemblance was produced by divergent rather than convergent evolution. In practice the two problems are attacked together, because no one is as interested in the more obvious (and highly significant) similarities that all proteins share simply as a result of globularity, covalent bonding, and preferred backbone conformations. Ideally one wants the "control" or reference comparisons to incorporate all nonhistorical constraints that apply to protein structure in general: requirements of overall stability, side chain packing, efficient folding, and all the other factors we do not yet know. Then any closer degree of resemblance can be assumed to be due to an historical evolutionary relationship (with a calculable confidence level).
The apparent objectivity of quantitative comparison methods obscures the fact that we do not yet know nearly enough about either the genetic processes or the stability and folding requirements to be sure the estimated probabilities of relationship are correct within an order of magnitude. Most comparison methods cannot readily allow for insertions and deletions; we know that they are an important factor that should be included, but even if the computational difficulties can be overcome, we simply do not have any idea of the relative likelihood of, for instance, one long versus two short insertions or of whether an insertion that makes a wide spatial excursion is any less likely than one which stays close. Because there are fewer degrees of freedom, spatial equivalence between helices must be less significant per residue than betweenβ strands than between nonrepetitive structure, but we cannot quantitate this effect. Functional resemblance is certainly a strong argument for the significance of a resemblance, but it cannot make a case for divergent rather than convergent evolution. Perhaps the most fundamental difficulty is that it is an a priori assumption, not an empirically determined fact, that closeness of spatial coordinates is a suitable measure of evolutionary distance.
At the same time, we need to know more about the genetic mechanisms that may be influential in protein evolution, since our current paradigms are almost certainly too simple. We need to understand more about the practical consequences of exon-intron organization on the gene and whether it generally correlates with domain divisions or with smaller internal units. It would be useful to know how unusual is the circularly permuted amino acid sequence of favin versus concanavalin A (Cunningham et al., 1979). And we might consider the possibility, for instance, that the helical portion of the larger domain of hexokinase (see Fig. 108) could "bud off" as an independent protein that would have an historical evolutionary relationship to the doubly wound sheet portion of hexokinase but no structural or sequence resemblance whatsoever. In the worst case, it could be that evaluating probable evolutionary relationships in terms of structural resemblance is not generally possible, because the constraints of stability and folding requirements might turn out to be more demanding than the limitations on rapid evolutionary change. However, one must start out with the more optimistic attitude that a sufficiently varied and open-minded program of structure comparisons will teach us a great deal both about the folding constraints and also about the evolutionary history of proteins.
It has been evident for some time both that a random search through all conformations could not possibly explain protein folding (Levinthal, 1968) and also that the structures themselves show evidence of systematic local folding patterns. The consistent presence of domains in the larger proteins strongly suggests that they are folding units (Gratzer and Beaven, 1969; Wetlauter, 1973), and for some proteins it is known experimentally that an isolated domain can fold spontaneously (e.g., Ghelis et al., 1978). A domain usually is made up from a single continuous portion of the backbone; however, the idea of separately folding domains, which then associate to form the intact protein, gains additional support from the frequency with which there occurs a short "tail" or "arm" at one end of a domain sequence which folds over to wrap against the outside of a neighboring domain. Figure 66 shows the structure of papain, which is a classic example of domain-clasping arms. Presumably, the placement of such arms is one of the last events in protein folding, which helps bind together the preformed domains.
It has frequently been pointed out (Wetlaufer, 1973; Ptitsyn and Rashin, 1975; Richardson, 1975; Levitt and Chothia, 1976) that the very high occurrence of associations between secondary-structure elements that are adjacent in the sequence is almost certainly a result of the fact that such nearest-neighbor elements are far more likely to come together during folding. This sort of regularity implies that at least some features of the final protein structure are under fairly strong control by the kinetic requirements of the folding process.
Additional sorts of regularities seen in our general classification of structures allow one to generalize the above idea still further. The prevalence of a few simple patterns of overall topology, and especially such features as the right-handedness of crossover connections and the frequency and handedness of Greek keys, strongly suggest the hypothesis that medium-sized as well as strictly local sections of polypeptide backbone have correlated conformations and tend to fold up as a concerted, interacting unit. One of the most interesting supports for this idea is the difference in statistical distribution of topologies that is seen between antiparallel α, antiparallel β, and parallel α/β structures. The parallel α/β structures are greatly influenced by the relatively long-range regularity of crossover handedness, which together with protection for both sides of the sheet produces the doubly wound α/β structure. In contrast to that situation, three-helix units with the first helix parallel to the third one show no handedness preference whatsoever. Although the possible topologies are exactly equivalent for antiparallel α bundles and for antiparallel β barrels, the frequency with which the various possibilities occur is very different for the two cases. Greek key topologies are about four times as common relative to up-and-down topologies for β structures as compared with α ones; the helical Greek keys occur with either handedness, while 12 of the 13 Greek key β barrels are counterclockwise. Pair associations in the β and α/β structures unambiguously show quite long-range correlations. Such correlations are most easily understood if fairly long portions of the polypeptide chain tend to fold as concerted units, such as the coiling up of a twisted, two-stranded β ribbon shown in Fig. 100. The distribution of features seen in helical proteins is ambiguous: it does not rule out the possibility of long-range concerted folding units, but it does not provide any particular support for such an idea. The observed helical structures could be explained by a simple model in which each new helix-pair association is independent of the topology of earlier pairs.
It has long been assumed that among the fluctuating conformational states early in the protein folding process, local elements of secondary structure are formed for a significant portion of the time, evidence comes from the experimentally observed behavior of synthetic polypeptides (e.g., Yaron et al., 1971), from theoretical calculations of locally determined stability (e.g., Ralston and DeCoen, 1974), and even from the degree of success achieved by secondary-structure predictions based only on single-residue, pair, or triplet sequences (e.g., Chou and Fasman, 1974). Particularly favorable such local regions of structure can act as nucleation sites to start and guide the folding process. Many proposed schemes of folding nucleation single out just one type of structure that seems especially suited for forming the first nuclei. The chief candidates that have been proposed as folding nuclei are α-helices, either alone (Levinthal, 1966; Anfinsen, 1972 and Lim, 1978) or in combination with β strands (Nagano, 1974); pairs of β strands brought together by a tight turn (Lewis et al., 1971; Crawford et al., 1973) or as long double ribbons (Ptitsyn and Finkelstein, 1980); and hydrophobic clusters (Matheson and Scheraga, 1978). The proposals for helical nuclei postulate that in predominantly β proteins the helices in the nucleation structures later unfold into extended strands. However, backbone connectivity has its maximum influence early in the folding process, so that topological patterns in the final structure are very sensitive to the order and mechanism of folding, as we have seen before in the contrast between the orderly topology of β strands and the random topology of disulfide connections. Therefore, if nucleation sites are basically similar for all types of structures, that similarity should show up in the overall topological patterns. Instead, as we have seen above, each of the broad types of structure shows characteristically different patterns of pair associations, coiled features, and handedness. Nucleation by hydrophobic clusters is harder to judge from the appearance of the final, folded structures. In proteins with strong long-range regularities of secondary structure it seems very unlikely that the earliest stages of folding are controlled entirely by hydrophobic associations, but there might be pure nucleation by hydrophobic clusters in the more irregular structures.
Judging from the types of regularities seen in the final structures, it seems likely that the typical folding nuclei are different for each of the three largest categories of structure: presumably those nuclei are individual helices and pairs of helices for the antiparallel α structures, β—α—β loops for the parallel α/β group, and two-stranded β ribbons for the antiparallel β structures. The small S—M proteins presumably either nucleate by helices or β ribbons which may be partially lost later or else by hydrophobic clusters. This diversity of folding nuclei would fit fairly well with Rose's "lines and hinges" model (Rose et al., 1976) except that different types of lines are not equivalent, and only for the antiparallel α case could they be considered as joined by completely flexible hinges. Tanaka and Scheraga (1977) have also proposed a model with diverse nuclei that are determined by neardiagonal regions of local interaction on the diagonal contact plot which fold by steps rather similar to the ones proposed below, except that forming contacts in rigorous order of increasing separation in the sequence does not permit explanation of any topological regularities larger than pairwise.
One last suggestive feature that is seen in the known protein structures is the frequency with which they "almost match" some prototypical structure. As an example of this sort of deviation, plastocyanin (Colman et al., 1978) is an antiparallel β barrel with seven well-formed β strands and an eighth strand which makes only one or two β-type hydrogen bonds, includes a short helix and an irregular excursion, and is slightly displaced from the position for an eighth β strand. If just the seven good β strands are counted as part of the barrel it has an unusual and complicated topology, but if the irregular eighth strand is included the structure is a Greek key barrel of the usual handedness. It may well be that plastocyanin folds as a more regular eight-stranded barrel but effectively loses the β structure in that eighth strand during the final process of adjustment to optimize fit for all the side chains. The significance of the eight-stranded Greek key structure for plastocyanin is reinforced by the fact that the Greek key structure is clearly present in the related protein azurin, with well-formed β structure for that same eighth strand (Adman et al., 1978). There are many other examples of such "approximate" pieces of structure although there is not always a convenient related protein to confirm the assignment. Such features could be explained if proteins first fold to form a maximum amount of regular secondary structure but then may lose some portions of the secondary structure in the final stage of adjusting all interactions for maximum stability. This sort of unfolding and loss of regularity at the final stages has been suggested before on varied sorts of evidence, both for helices (Carter et al., 1974; Lim, 1978) and for β strands (Ko et al., 1977; Richardson et al., 1978). The entire category of S—M proteins is presumably an exaggerated case of this sort of process, in which the amount of adjustment needed to accommodate disulfides or metals into these small proteins is often enough to disrupt the secondary structure almost beyond recognition.
By putting together all of the ideas discussed above, we can propose a speculative general scheme of protein folding as suggested by the properties of the final structures.
The proposed folding process involves four stages, which could be expected to be at least partially separated in time but are not rigorously sequential. Figure 109 illustrates the stages of folding as they might apply to each of the major structure categories. The first stage is the classic one of forming, in a probabilistic and fluctuating sense, individual elements of α-helix, extended strand, or tight turns and of combining two or three of those elements into the first folding nuclei. This does not involve backbone conformations different from those that would be present in a rigorously random coil; it simply involves a difference in the statistics of their distribution in favor of more correlation between the conformations of adjacent residues. Helices have the advantage of hydrogen-bond formation and of cooperativity, and the helices undoubtedly are more regular and can persist for much longer times than isolated, or perhaps even than paired β strands. However, extended strands have the advantage that a much broader range of conformational angles is capable of taking part in β structure, and it could well be that extended strands capable of further interaction are present for about as large a fraction of the time as are individual helices. Once a pair of helices, β—α—β loop, a two-stranded β ribbon, or a large hydrophobic cluster has formed, it would presumably have enough stability to act as a nucleation site for further folding. At least for large domains, it seems unlikely that there is a unique initial folding nucleus, since the relative stability and probability of occurrence would often be similar among, for instance, several possible β—α—β loops. Indeed, the most common topologies are the ones that would permit the most alternative folding pathways (Richardson, 1977).
The second stage of folding is the growth and coalescence of secondary-structure elements two or three at a time to form successively larger substructures. The characteristic associations formed at this stage depend on the type and order of secondary-structure elements in the sequence. All-helical structures may associate fairly independently, one nearest-neighbor pair at a time. It is proposed that β—α—β structures fold concertedly by throwing up loops. Antiparallel β structures probably form two-stranded ribbons from nearest-neighbor strands separated by turns; they can then add on strands or pairs of strands to either side of an initial ribbon, or they can coil up a very long ribbon into a Greek key. At the end of this second stage all of the major regular structures are in place, sometimes in a more complete or more regular form than in the final native structure.
The third stage is a process of many readjustments to settle down into a comfortable, stable overall structure. At this stage disulfides are joined in their final native pairing, metals and prosthetic groups are bound, β bulges are formed, and cis-trans isomerization of prolines occurs if necessary (see Brandts et al., 1975, 1977). Side chain conformations are adjusted to provide optimal fit, and some main chain conformations are also adjusted. Occasionally this might produce additional secondary-structure interactions, but it is much more likely to disrupt some of the preexisting secondary structure; mainchain hydrogen-bonding lost at this stage is more than compensated by side chain interactions. This third, readjustment, stage of folding would normally be expected to be very much slower than any of the other steps. For a one-domain, single subunit protein the folding process would then be complete (unless proteolytic cleavages or some other modifications are needed). It may be that the kind of major reshuffling seen during the folding of pancreatic trypsin inhibitor (Creighton, 1977) can be considered as an especially pronounced example of these final readjustments, although the fact that the incorrect intermediates are not very compact suggests that they may represent a rather different process that can happen in addition to the steps considered here. In general, the final structures of the small S—M proteins suggest that they undergo more extensive rearrangement than other proteins.
The fourth stage of folding is the association of domains (and/or subunits). Sometimes association might start at the end of the second stage, but in general it would probably happen only after readjustments within domains were fairly complete. Domains primarily associate as rigid bodies, but there are usually adjustments of side chains at the contact surface, and "arms" that clasp opposite domains cannot old mto their final conformation until this last stage. Association of subunits is equivalent to association of domains, except for the difference in kinetics produced by the covalent attachment.
The most characteristic features of this proposed folding scheme are the proposal of different kinds of nucleation for the different major structure types, the postulation of some rather large-scale concerted folding units, and the prediction of folding intermediates with somewhat greater amounts of the same sort of secondary structure found in the final nahve protein. The last effect might turn out to be most pronounced in those proteins with very irregular secondary structures.
In the final analysis protein folding will be really understood only with the aid of much more extensive, direct experimental evidence. Speculatlve hypotheses can be useful, however, in suggesting potentially fruitful questions for experimental investigation. Probably the most important idea suggested by the above schema is that there are likely to be considerable systematic differences in the kinetics of foldmg between the various major structural categories of proteins.
I am especially grateful to David Richardson for, among other things, the meticulous technical photography; to Richard Feldmann for extensive use of his molecular display system; and to Chris Anfinsen for suggesting that this article be written.
I am also greatly indebted to the following people, who provided unpublished coordinates or other information: Pat Argos, Frances Bernstein, Colin Blake, David Blow, Tom Blundell, Rick Bott, Carl Brändén, John Chambers, David Davies, Phil Evans, Alyosha Finkelstein, Bob Fletterick, Hans Freeman, Irving Geis, Pauline Harrison, Steve Harrison, Wayne Hendrickson, Isabella Karle, Aaron Klug, Joe Kraut, Michaei Lelbman, Anders Liljas, Bill Lipscomb, Martha Ludwig, Scott Mathews, Brian Matthews, Alex McPherson, Hilary Muirhead, George Némethy, Eduardo Padlan, Oleg Ptitsyn, Michael Rossmann, Ray Salemme, Charlotte Schellman, George Schulz, Christine Slingsby, Tom Steitz, Michael Sternberg, Bob Stroud, Martha Teeter, Janet Thornton, Al Tulinsky, B.K. Vainshtein, Pat Weber, Don Wetlaufer, Don Wiley, Alex Wlodawer, and Christine Wright.
This work was supported by NIH grant GM-15000.